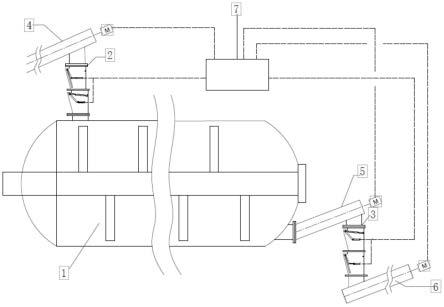
1.本发明属于晶型药物领域,具体涉及一种硫酸阿扎那韦晶型的生产方法。
背景技术:
2.艾滋病(aids),又称获得性免疫缺陷综合征,是由人类免疫缺陷病毒(human immunodeficiency virus,简称hiv)特异性感染和杀伤人体主要淋巴细胞所导致的免疫缺陷疾病。个体感染后由于免疫系统被破坏,易发生机会性感染和恶性肿瘤,病死率较高,现阶段尚无彻底治愈的药物和方法(参见:白跃飞,刘举,周宏,等.hiv蛋白酶抑制剂硫酸阿扎那韦的原料药合成研究进展:中国新药杂志,2018年,第27卷第7期:767-780)。
3.目前,感染者主要通过hiv蛋白酶抑制剂、逆转录酶抑制剂、整合酶抑制剂等药物进行治疗,以此来提高患者的生活质量和有效寿命。其中,阿扎那韦是21世纪初研制成功的一种该类药物。因硫酸阿扎那韦在动物体内具有比游离碱(即:阿扎那韦)更高的口服生物利用度,并且比其他盐具有更好的水中溶解度和稳定性,所以在实际应用中常常以硫酸阿扎那韦的形式进行给药。
4.硫酸阿扎那韦,也称为阿扎那韦硫酸盐或阿扎那韦硫酸氢盐,化学名称:(3s,8s,9s,12s)-3,12-双(1,1-二甲基乙基)-8-羟基-4,11-二氧代-9-(苯甲基)-6-[[4-(2-嘧啶)苯基]甲基]-2,5,6,10,13-五氮杂十四烷二酸二甲酯硫酸氢盐,分子式:c
38h52
n6o7·
h2so4,其结构式如下:
[0005][0006]
因其药效强,能够持续强效地抑制hiv病毒,并具有低耐药、用药方便、不良反应小等优点,已逐步成为现阶段最具潜力的一线治疗艾滋病的药物。
[0007]
已有相关文献报道,硫酸阿扎那韦存在包括水合物、溶剂合物在内的多种晶型形式,主要有i型(后被称为a型或晶型a)、ii型、c型(也称为c模式)、e3型、h1型等;其中,a型为无水、非溶剂化晶型,ii型为水合吸湿性晶型,c型为水合物晶型,e3型为三乙醇溶剂合物晶型。
[0008]
在这些已知的晶型中,晶型a是一种比较重要的晶型形式,受到人们较多的关注。原研百时美-施贵宝控股爱尔兰无限公司,是通过以下方法来制备硫酸阿扎那韦晶型a:使阿扎那韦游离碱溶液与浓硫酸反应,所加入量的浓硫酸可与少于约15%重量的游离碱反应,然后将硫酸阿扎那韦a型晶种加入反应混合物中,当硫酸氢盐晶体形成时,根据三次方
程分多阶段以增加的速率加入另外的浓硫酸(也被称为:三次结晶技术或改良的三次结晶技术),最终才能有效形成阿扎那韦硫酸氢盐的a型晶体;而如果用乙醇作为反应溶剂和庚烷作为析晶溶剂,通常情况下得到的是硫酸阿扎那韦e3型:三乙醇溶剂合物(参见:cn1980666a和cn101565398a)。
[0009]
由于该原研方法在滴加硫酸时需采用三次方程多阶段且增加速率的滴加方式,操作要求繁琐和苛刻,且不易控制,不利于工业化生产。
[0010]
为了解决这一技术问题,cn 105859611 a提供了一种制备阿扎那韦硫酸氢盐a型结晶的改进方法,具体做法是:a)将阿扎那韦游离碱置于乙醇中,室温下搅拌,然后滴加浓硫酸,将反应液加热搅拌,再加入惰性溶剂,冷却结晶,过滤后干燥,得到阿扎那韦乙醇合物e型结晶;b)将步骤a)得到的阿扎那韦乙醇合物e型结晶置于丙酮中,加热回流搅拌,冷却过滤后干燥,得到阿扎那韦硫酸氢盐a型结晶。
[0011]
也就是说,cn 105859611 a的方法是先使阿扎那韦游离碱在乙醇中和硫酸反应得到阿扎那韦乙醇合物e型结晶,再通过转晶,最终才得到阿扎那韦硫酸氢盐a型结晶。虽然该方法相对于原研方法而言有所改进,但仍然稍显繁琐:需要先制备得到阿扎那韦乙醇合物e型结晶,再经过转晶,才能最终得到硫酸阿扎那韦晶型a,也不是十分利于产业化生产。
[0012]
有鉴于此,特提出本发明。
技术实现要素:
[0013]
针对现有技术存在的问题和/或不足,本发明的目的在于提供一种硫酸阿扎那韦晶型的生产方法。该方法,能够很方便地控制所得产物硫酸阿扎那韦的晶型形式,与现有技术相比,大大提升了生产、操作和/或控制的便捷性,更有利于产业化生产,显著提高硫酸阿扎那韦晶型药物特别是硫酸阿扎那韦晶型a的可及性。
[0014]
本发明提供的一种硫酸阿扎那韦晶型的生产方法,它包括以下步骤:使用或不使用惰性气体保护,阿扎那韦与硫酸在乙醇中反应后,加入正庚烷,结晶,分离,干燥,得到硫酸阿扎那韦晶型;
[0015]
其中,乙醇用量是阿扎那韦重量的3.5~15倍,和/或,正庚烷用量是阿扎那韦重量的2.5~12倍。
[0016]
进一步的,
[0017]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型a时,乙醇用量是阿扎那韦重量的6.4~6.8倍,和/或,正庚烷用量是阿扎那韦重量的5.8~6.2倍。
[0018]
进一步的,
[0019]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型a时,乙醇用量是阿扎那韦重量的6.6倍,和/或,正庚烷用量是阿扎那韦重量的6倍。
[0020]
进一步的,
[0021]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型a时,乙醇与正庚烷的重量比为1∶0.88~0.95;优选的,乙醇与正庚烷的重量比为1∶0.9~0.92。
[0022]
进一步的,
[0023]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型a时,反应温度为10~30℃,和/或,结晶温度为10~30℃;优选的,反应温度为25℃
±
2℃,和/或,结晶温度为25℃
±
2℃。
[0024]
进一步的,
[0025]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型e3时,乙醇用量是阿扎那韦重量的3.6~4倍,和/或,正庚烷用量是阿扎那韦重量的2.5~3.2倍;优选的,乙醇用量是阿扎那韦重量的3.8倍,和/或,正庚烷用量是阿扎那韦重量的3倍。
[0026]
进一步的,
[0027]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型e3时,乙醇与正庚烷的重量比为1∶0.7~0.8;优选的,乙醇与正庚烷的重量比为1∶0.78~0.8。
[0028]
进一步的,
[0029]
当所述的硫酸阿扎那韦晶型为硫酸阿扎那韦晶型e3时,反应温度为32~40℃,和/或,结晶温度为32~40℃;优选的,反应温度为35℃
±
2℃,和/或,结晶温度为35℃
±
2℃。
[0030]
进一步的,
[0031]
在上述任一所述硫酸阿扎那韦晶型的生产方法中,阿扎那韦与硫酸的摩尔比为1∶1~1.5,和/或,结晶析出的时间为15~30h;优选的,阿扎那韦与硫酸的摩尔比为1∶1.08~1.12,和/或,结晶析出的时间为18~24h。
[0032]
本发明还提供了一种硫酸阿扎那韦晶型h1的生产方法,其特征在于,它包括以下步骤:按照上述任一所述硫酸阿扎那韦晶型的生产方法,得到硫酸阿扎那韦晶型a或硫酸阿扎那韦晶型e3,再按照已知方法,进行转晶,得到硫酸阿扎那韦晶型h1。
[0033]
本发明的有益效果是:本发明另辟蹊径,通过采用与现有技术截然不同的技术手段:调整反应溶剂(乙醇)和析晶溶剂(正庚烷)的用量及配比,意外地发现,该方法可以很方便地控制所得产物硫酸阿扎那韦的晶型形式,大大地提升了生产、操作和/或控制的便捷性,从而可以更方便、更容易地得到硫酸阿扎那韦晶型a,也更有利于产业化生产,显著提高硫酸阿扎那韦晶型药物的可及性,进而为保障我国医药产业持续健康发展不断地添砖加瓦。
[0034]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
附图说明
[0035]
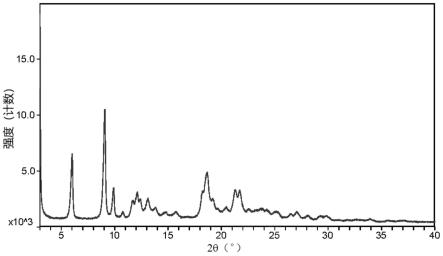
图1为实施例3所得产物的x-射线粉末衍射(xrd)图。
[0036]
图2为实施例8所得产物的x-射线粉末衍射(xrd)图。
具体实施方式
[0037]
下面将结合具体实施例对本发明进行清楚、完整的描述,本领域技术人员将会理解,下面所述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为对本发明保护范围的限制。
[0038]
本发明中,未注明具体条件者,按照常规条件或制造商建议的条件进行,所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0039]
关于本发明中使用术语的定义,除非另有说明,本文中术语提供的初始定义适用于全文中的该术语;对于本文没有具体定义的术语,应当根据公开内容和/或上下文,给出本领域技术人员能够给予它们的含义。
[0040]
实施例1
[0041]
氮气保护下,向反应釜中加入30kg(约42.56mol)阿扎那韦和100kg无水乙醇,搅拌均匀,加入4.8kg浓硫酸(约含46.5mol的h2so4),控制体系温度为35℃
±
2℃,搅拌2h,过滤,用14kg无水乙醇淋洗滤饼,在所得滤液中加入90kg正庚烷,35℃
±
2℃搅拌析晶20h,离心,用无水乙醇-正庚烷(质量比1∶0.9)的混合溶剂淋洗滤饼,真空干燥(真空度≤-0.09mpa),得到硫酸阿扎那韦晶型e3(三乙醇溶剂合物),hplc纯度99.5%,最大单杂0.31%,收率82.38%;经xrd检测,所得产物的xrd图基本上与cn 1980666a(发明名称:制备阿扎那韦硫酸氢盐的方法和新的形式,申请日:2005年5月3日)中所记载晶型e3的特征峰一致。
[0042]
实施例2
[0043]
氮气保护下,向反应釜中加入30kg(约42.56mol)阿扎那韦和350kg无水乙醇,搅拌均匀,加入4.8kg浓硫酸(约含46.5mol的h2so4),控制体系温度为35℃
±
2℃,搅拌2h,过滤,用25kg无水乙醇淋洗滤饼,将所得滤液35℃
±
2℃搅拌析晶20h,离心,用无水乙醇淋洗滤饼,真空干燥(真空度≤-0.09mpa),得到硫酸阿扎那韦晶型e3(三乙醇溶剂合物);经xrd检测,所得产物的xrd图基本上与cn 1980666a中所记载晶型e3的特征峰一致。
[0044]
实施例3
[0045]
氮气保护下,向反应釜中加入30kg(约42.56mol)阿扎那韦和178kg无水乙醇,搅拌均匀,加入4.84kg浓硫酸(约含46.88mol的h2so4),控制体系温度为25℃
±
2℃,搅拌2h,过滤,用20kg无水乙醇淋洗滤饼,在所得滤液中加入180kg正庚烷,25℃
±
2℃搅拌析晶20h,离心,用无水乙醇-正庚烷(质量比1∶0.9)的混合溶剂淋洗滤饼,真空干燥(真空度≤-0.09mpa),得到硫酸阿扎那韦,为白色固体,hplc纯度99.89%,最大单杂0.05%,收率84.55%。
[0046]
ir结果显示所得产物的红外吸收光谱与硫酸阿扎那韦标准品的红外吸收光谱主要特征吸收峰一致;hplc结果显示所得产物主峰保留时间与标准品主峰保留时间一致。
[0047]
对所得产物进行x-射线粉末衍射(xrd)检测,测试条件如下:
[0048]
设备型号:德国bruker d8 advance;cu(40kv,40ma);计算晶格间距d的波长=(cu/k-alphal)。
[0049]
检测结果见表1和图1。
[0050]
表1、实施例3所得产物的x-射线粉末衍射(xrd)检测结果
[0051][0052]
xrd结果表明,所得产物硫酸阿扎那韦在衍射角2θ=6.02、9.08、9.90、11.72、18.22、18.67(
±
0.2)度处有特征峰,与cn 1980666a中所记载晶型a的特征峰是一致的,从而证实实施例3所得产物是硫酸阿扎那韦晶型a。
[0053]
实施例4
[0054]
与实施例3相同的内容不再重复,不同的是,将浓硫酸的用量变更为4.4kg(约含42.62mol的h2so4),即:阿扎那韦与硫酸(h2so4)的摩尔比为1∶1,再按照相同的工艺条件,制备得到硫酸阿扎那韦,hplc纯度98.56%,收率77.5%;经xrd检测,所得产物的xrd图基本上与cn 1980666a中所记载晶型a的特征峰一致。
[0055]
实施例5
[0056]
与实施例3相同的内容不再重复,不同的是,将浓硫酸的用量变更为5.28kg(约含51.14mol的h2so4),即:阿扎那韦与h2so4的摩尔比为1∶1.2,再按照相同的工艺条件,制备得到硫酸阿扎那韦,hplc纯度99.92%,收率69.7%;经xrd检测,所得产物的xrd图基本上与cn 1980666a中所记载晶型a的特征峰一致。
[0057]
实施例6
[0058]
与实施例3相同的内容不再重复,不同的是,将搅拌析晶的时间从20h调整至23h,再按照相同的工艺条件,制备得到硫酸阿扎那韦,hplc纯度99.85%,收率82.38%;经检测所得产物的xrd图基本上与cn 1980666a中所记载晶型a的特征峰一致。
[0059]
实施例7
[0060]
以实施例1所得的硫酸阿扎那韦晶型e3(三乙醇溶剂合物)为原料,按照专利cn107382833a公开的已知方法(如:实施例8),进行转晶,得到硫酸阿扎那韦晶型h1。
[0061]
实施例8
[0062]
以实施例3所得的硫酸阿扎那韦晶型a为原料,按照专利cn 107382833 a公开的已知方法(如:实施例5),进行转晶,得到产物:硫酸阿扎那韦晶型h1,其xrd检测结果见表2和图2。
[0063]
表2、实施例8所得产物的x-射线粉末衍射(xrd)检测结果
[0064][0065]
此外,
[0066]
本发明的研究以及生产实践还发现,与现有技术的相关报道一致:以乙醇作为反应溶剂和庚烷作为析晶溶剂,在大多数情况下都得到的是硫酸阿扎那韦晶型e3(三乙醇溶剂合物)或者是混晶,而只有在特定的用量及配比的较窄范围条件下得到的产物才是硫酸阿扎那韦晶型a;而且,结晶温度对所得产物的晶型形式也有较大影响,在10~30℃的温度范围内,对所得产物是硫酸阿扎那韦晶型a有利,而结晶温度超过30℃(如:35℃
±
2℃),往往得到的是硫酸阿扎那韦晶型e3(三乙醇溶剂合物)或者混晶。
[0067]
当然,本发明还可以有其它多种实施方式,在不违背本发明精神及其实质的情况下,熟悉本领域的技术人员可以根据本发明作出各种相应的改变和/或变形,这些相应的改变和/或变形都应属于本发明所附的权利要求的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。