1.本发明涉及化学合成技术领域,特别涉及一种瑞德西韦的合成方法。
背景技术:
2.瑞德西韦(remdesivir),是吉利德化学在研药品,属于核苷类似物,是rna依赖的rna聚合酶抑制剂,可以通过抑制病毒核酸合成抗病毒。目前针对埃博拉病毒感染的临床研究进行到了ii期阶段。感染mers的小鼠接受这种联合疗法后表现要好得多,病毒复制减少,肺功能改善。近期研究表明,瑞德西韦对抑制新冠病毒有一定活性作用。
3.临床试验上用于治疗埃博拉病毒感染,结果疗效非常好。随后研究发现,瑞德西韦不仅对埃博拉病毒这类丝状病毒有效,对于呼吸道合胞病毒、冠状病毒、尼帕病毒以及亨德拉病毒也有抑制效果。此外,经体外细胞实验和动物模型实验证实瑞德西韦对非典型性肺炎冠状病毒和中东呼吸综合症冠状病毒均产生抗病毒作用。虽然目前没有抗病毒数据显示瑞德西韦对于2019
‑
ncov的活性,且该药物尚未在全球任何地区获得许可或批准,其安全性和有效性也未被证实;但是该药物针对sars
‑
cov和mers
‑
cov的有效数据给了我们治疗新型病毒的参考。
4.瑞德西韦化学名:(s)
‑2‑
乙基丁基
‑2‑
(((r)
‑
((2r,3s,4r,5r)
‑5‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑5‑
氰基
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲氧基)(苯氧基)磷酰基)氨基丙酸酯,化学结构式如下:,现有技术中与合成瑞德西韦有关的中间体主要包括以下几个:中间体1化学名:(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈,化学结构式如下:,中间体2化学名:(s)
‑2‑
乙基丁基2
‑
(((s)
‑
((2r,3s,4r,5r)
‑5‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑5‑
氰基
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲氧基)(苯氧基)磷酰基)氨基丙酸酯,化学结构式如下:,中间体3化学名:(2s)
‑2‑
乙基丁基2
‑
((((2r,3s,4r,5r)
‑5‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑5‑
氰基
‑
3,4
‑
二羟基四氢呋喃
‑2‑
基)甲氧基)(苯氧基)磷酰基)氨基丙酸酯,化学结构式如下:,中间体4化学名:(3ar,4r,6r,6ar)
‑4‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑6‑
(羟甲基)
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二恶英
‑4‑
碳腈,化学结构式如下:,中间体5化学名:(s)
ꢀ‑2‑
乙基丁基
‑2‑
((r)
‑
((3ar,4r,6r,6ar)
‑6‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑6‑
氰基
‑
2,2
‑
二甲基四氢呋喃[3,4
‑
d][1,3]二氧基
‑4‑
甲氧基)(苯氧基)磷酰)氨基丙酸酯,化学结构式如下:
,中间体6化学名:(s)
ꢀ‑2‑
乙基丁基
‑2‑
((s)
‑
(4
‑
硝基苯氧基)(苯氧基)磷酰基)氨基丙酸酯,化学结构式如下:,中间体7化学名:(s)
ꢀ‑2‑
乙基丁基
‑2‑
((s)
‑
(全氟苯氧基)(苯氧基)磷酰基)氨基丙酸酯,化学结构式如下:。
[0005]
目前国内外由中间体1合成至瑞德西韦主要有以下两条路线:journalofmedicinalchemistry2017年60卷1648
‑
1661页公开了中间体1至瑞德西韦的第一代合成方法,合成路线如下,直接合成得到的瑞德西韦是消旋体,需要高效液相色谱法进行分离才能得到单一构型化合物,收率极低,不适合工业放大;
;
②
nature杂志2016年531卷381
‑
385页报道了瑞德西韦的改进合成方法,合成路线如下,中间体1经丙酮叉保护双羟基合成中间体4,再与中间体6完成对接反应,最后水解得到最终产物瑞德西韦。三步反应的收率分别为90%、70%和69%,总收率仅为43%,原料成本相对较高,合成中间体4时,使用浓硫酸较危险,后处理繁琐,生产周期长,生产效率低,废液量大,大大增加了工业生产成本,而且该方法使用了有基因毒性的硝基取代物中间体6,增加了出现基因毒杂质的风险。
[0006]
。
技术实现要素:
[0007]
为了解决上述背景技术中提出的问题,本发明提供一种瑞德西韦的合成方法。
[0008]
本发明提供的一种瑞德西韦的合成方法,包括以下步骤:、将(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈、2,2
‑
二甲氧基丙烷和第一酸催化剂加入第一溶剂中,搅拌,经2,2
‑
二甲氧基丙烷保护邻二羟基生成中间体4,反应完毕后调碱降温;所述中间体4的合成路线如下:
;、将无水氯化镁和中间体7加入步骤(1)合成的中间体4中,通氮气流保护,搅拌均匀后滴加碱催化剂,升温搅拌,经对接反应合成中间体5,反应完毕,进行提取分液;所述中间体5的合成路线如下:;、向步骤(2)合成的中间体5中滴加第二酸催化剂,搅拌,水解反应完毕后,经过后处理得到瑞德西韦粗品;所述瑞德西韦粗品的合成路线如下:。
[0009]
优选的,所述步骤(1)中第一酸催化剂为硫酸、甲磺酸、磷酸、乙酸和对甲苯磺酸中的一种或几种,第一溶剂为二氯甲烷、乙酸乙酯、四氢呋喃、丙酮、乙腈、甲醇和乙醇中的一种,反应温度为10~25℃,搅拌时间为4~6h,所述调碱为滴加三乙胺、n,n
‑
二异丙基乙胺和碳酸钾水溶液中的一种或几种,保持滴加温度为10℃,直至ph为8~9,调碱完毕后降温至0~10℃,所述(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈、2,2
‑
二甲氧基丙烷和第一酸催化剂的物质的量之比为1:(5~8):(1.05~1.5),(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈与第一溶剂的质量比为1:(5~10)。
[0010]
优选的,所述步骤(2)的碱催化剂为氢氧化钾、氢氧化钠、碳酸钾、氢化钠、三乙胺、二乙胺和n,n
‑
二异丙基乙胺中的一种或几种,滴加碱催化剂的温度为0~10℃,反应温度为10~25℃,搅拌时间为2~4h,所述提取分液的具体步骤为:向反应中滴加10%柠檬酸水溶液,保持温度为0~10℃,直至ph为3~4,分液,水相弃掉,有机相中滴加20%碳酸钾水溶液,保持温度为10℃,直至ph为8~9,分液,水相弃掉,有机相降温至0~10℃。
[0011]
优选的,所述(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈、中间体7、无水氯化镁和碱催化剂的物质的量之比为1:(1.1~1.5):(1.1~2.0):(3~5)。
[0012]
优选的,所述步骤(3)中第二酸催化剂为质量分数为36%的浓盐酸或乙酸,滴加第二酸催化剂的温度为0~10℃,反应温度为0~10℃,搅拌时间为6~8h。
[0013]
优选的,所述(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈与第二酸催化剂的物质的量之比为1:(5~9)。
[0014]
优选的,所述步骤(3)中的后处理的具体步骤为:向反应中滴加20%碳酸钾水溶液,保持温度为
‑
5~5℃,直至ph为8~9,30~40℃浓缩有机相,浓缩完毕后加入第二溶剂,环境温度滴加纯化水至大量固体析出,降温至0~10℃,搅拌6~8h,过滤得到瑞德西韦粗品。
[0015]
优选的,所述第二溶剂为二氯甲烷、乙酸乙酯、四氢呋喃、丙酮、乙腈、甲醇和乙醇中的一种或几种,所述(2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈、第二溶剂和纯化水的质量比为1:8:15,所述环境温度为20~30℃。
[0016]
优选的,所述合成方法还包括对所述瑞德西韦粗品进行精制的步骤,所述精制包括将所述步骤(3)制备的瑞德西韦粗品加入第三溶剂中,搅拌加热至完全溶解,停止加热,滴加纯化水,降温析晶,离心,真空干燥。
[0017]
优选的,所述第三溶剂为甲醇、乙醇和异丙醇中的一种或几种,所述瑞德西韦粗品、第三溶剂和纯化水的质量比为1:5:10,所述搅拌加热的温度为50~80℃,降温析晶的温度为0~10℃,搅拌时间为6~8h,真空干燥的温度为40~60℃,压力为0.7~0.9mpa,真空干燥的时间为8~12h。
[0018]
本发明所用的中间体7采购自山东诚汇双达药业有限公司。
[0019]
综上所述,本发明具有如下的有益技术效果:本发明的三步反应均以第一溶剂为介质进行反应,仅在三步反应完成后进行后处理浓缩溶剂,减少浓缩溶剂的次数,降低工业成本;且反应条件温和、操作简单、副反应少、生产周期短,废液量大大减少,减轻环保压力,方便溶剂回收,大大节约了生产成本,中间体无过多后处理损失;获得产品的收率高、纯度高,特别适用于工业化生产,对药品质量控制与临床疗效具有意义。
附图说明
[0020]
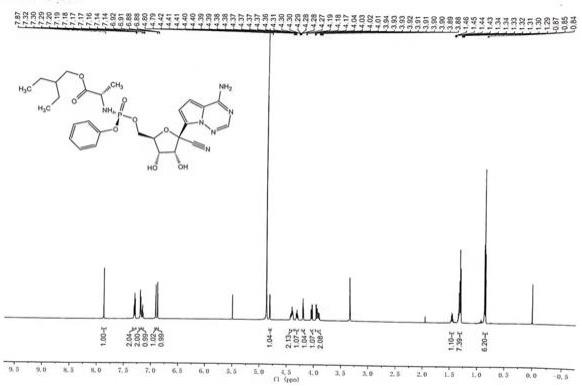
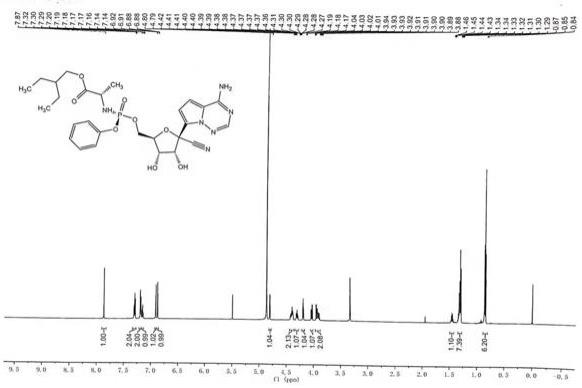
图1是本发明实施例5的高效液相色谱图;图2是本发明实施例5的氢谱图。
具体实施方式
[0021]
以下结合实施例对本发明作进一步详细说明。
[0022]
实施例1向250ml反应瓶中加入50g第一溶剂二氯甲烷、5g(17.2mmol) (2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈和8.9 g(85.5 mmol)2,2
‑
二甲氧基丙烷,将1.77g(18.06mmol)第一酸催化剂硫酸慢慢加入反应瓶
中,在25℃下搅拌4h,反应完毕后滴加三乙胺调碱,保持滴加温度为10℃,直至ph为9,调碱完毕后降温至8℃;将11.1g(22.4mmol)中间体7和2.6 g(27.5mmol)无水氯化镁加入反应瓶中,真空通氮气置换三次,搅拌均匀后缓慢滴加8.7g(86mmol)碱催化剂三乙胺,滴加过程中温度控制在10℃,滴加完毕后升温到25℃,继续搅拌4h,反应完毕后,向反应中滴加10%柠檬酸水溶液,保持温度为10℃,直至ph为4,分液,水相弃掉,有机相中滴加20%碳酸钾水溶液,保持温度为10℃,直至ph为9,分液,水相弃掉,有机相降温至10℃;向反应中滴加13.76g (137.6 mmol)第二酸催化剂36%的浓盐酸,温度控制在10℃,搅拌反应8h,反应完毕后,滴加20%碳酸钾水溶液,保持温度在5℃,直至ph为9,30℃浓缩有机相,浓缩完毕后加入40g第二溶剂二氯甲烷,以便后续析晶。环境温度20℃滴加75g纯化水至大量固体析出,降温至0℃搅拌6h,过滤得到瑞德西韦粗品。收率:76%,纯度:90%。
[0023]
实施例2向250ml反应瓶中加入400g第一溶剂乙酸乙酯、50g(172mmol) (2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈和106.8 g(1032mmol)2,2
‑
二甲氧基丙烷,将18.1g(189mmol)第一酸催化剂甲磺酸慢慢加入反应瓶中,在10℃下搅拌6h,反应完毕后滴加n,n
‑
二异丙基乙胺调碱,保持滴加温度为10℃,直至ph为8,调碱完毕后降温至0℃;将93.9g(189mmol)中间体7和29.25g(344mmol)无水氯化镁加入反应瓶中,真空通氮气置换三次,搅拌均匀后缓慢滴加50.2g (688mmol)碱催化剂二乙胺,滴加过程中温度控制在0℃,滴加完毕后升温到10℃,继续搅拌2h,反应完毕后,向反应中滴加10%柠檬酸水溶液,保持温度为0℃,直至ph为3,分液,水相弃掉,有机相中滴加20%碳酸钾水溶液,保持温度为0℃,直至ph为8,分液,水相弃掉,有机相降温至0℃;向反应中滴加72.3g (1204mmol)第二酸催化剂乙酸,温度控制在0℃,搅拌反应6h,反应完毕后,滴加碳酸钾水溶液,保持温度在
‑
5℃,直至ph为8,40℃浓缩有机相,浓缩完毕后加入400g第二溶剂乙酸乙酯,以进行后续析晶。环境温度30℃滴加750g纯化水至大量固体析出,降温至10℃搅拌8h,过滤得到瑞德西韦粗品。收率:78%,纯度:93%。
[0024]
实施例3向200l反应釜中加入35kg第一溶剂四氢呋喃、5kg(17.2mol) (2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈和12.46 kg (120.4mol)2,2
‑
二甲氧基丙烷,将1.3kg (22.4mol) 第一酸催化剂乙酸慢慢加入反应釜中,在15℃下搅拌5h,反应完毕后滴加三乙胺,保持滴加温度为10℃,直至ph为9,调碱完毕后降温至5℃,将12.8kg(25.8mol)中间体7和2.275 kg (24.08mol)无水氯化镁加入反应瓶中,真空通氮气置换三次,搅拌均匀后缓慢滴加6.67kg (51.6mol)碱催化剂n,n
‑
二异丙基乙胺,滴加过程中温度控制在5℃,滴加完毕后升温到15℃,继续搅拌3h,反应完毕后,向反应中滴加10%柠檬酸水溶液,保持温度为5℃,直至ph为4,分液,水相弃掉,有机相中滴加20%碳酸钾水溶液,保持温度为5℃,直至ph为9,分液,水相弃掉,有机相降温至5℃;向反应中滴加15.5 kg(154.8mol)第二酸催化剂36%的浓盐酸,温度控制在5℃,搅拌反应7h,反应完毕后,滴加碳酸钾水溶液,保持温度在0℃,直至ph为9,35℃浓缩有机相,浓缩完毕后加入40kg第二溶剂四氢呋喃,环境温度25℃滴加75kg纯化水至大量固体析出,降温至5℃搅拌7h,过滤得到瑞德西韦粗品。收率:75%,纯度:92%。
[0025]
实施例4
向1000l反应釜中加入125kg第一溶剂丙酮、25kg(86mol) (2r,3r,4s,5r)
‑2‑
(4
‑
氨基吡咯[2,1
‑
f][1,2,4]三嗪
‑7‑
基)
‑
3,4
‑
二羟基
‑5‑
(羟甲基)四氢呋喃
‑2‑
碳腈和71.2 kg (688mol)2,2
‑
二甲氧基丙烷,将12.6kg (129mol)第一酸催化剂磷酸慢慢加入反应釜中,在20℃下搅拌6h,反应完毕后滴加碳酸钾水溶液,保持滴加温度为10℃,直至ph为8,调碱完毕后降温至7℃,将60.1kg(103.2mol)中间体7和8.94kg (94.6mol)无水氯化镁加入反应瓶中,真空通氮气置换三次,搅拌均匀后缓慢滴加41.5kg(301mol)碱催化剂碳酸钾,滴加过程中温度控制在6℃,滴加完毕后升温到20℃,继续搅拌2.5h,反应完毕后,向反应中滴加10%柠檬酸水溶液,保持温度为7℃,直至ph为3,分液,水相弃掉,有机相中滴加20%碳酸钾水溶液,保持温度为7℃,直至ph为8,分液,水相弃掉,有机相降温至7℃;向反应中滴加25.8kg (430mol)第二酸催化剂乙酸,温度控制在6℃,搅拌反应6.5h,反应完毕后,滴加碳酸钾水溶液,保持温度在3℃,直至ph为8,38℃浓缩有机相,浓缩完毕后加入200kg丙酮,环境温度30℃滴加375kg纯化水至大量固体析出,降温至8℃搅拌7h,过滤得到瑞德西韦粗品。收率:81%,纯度:92%。
[0026]
实施例5将1 kg瑞德西韦粗品(实施例4)加入5 kg第三溶剂甲醇中,搅拌加热至50
°
c溶解完全,停止加热,继续搅拌,滴加10kg纯化水,降温至0 ℃,搅拌8h,离心,在40℃,0.9mpa的条件下真空干燥8h,得到900 g瑞德西韦精制品。收率:90 %,高效液相色谱法检测纯度,色谱图见图1,经检测纯度:99.97%,单杂均小于0.1 %,氢谱图见图2,对氢谱图的分析如下:6.88 ppm
‑
7.87 ppm为芳香环上的氢,共8个;3.86 ppm
‑
4.80 ppm为糖环以及与酯基和磷酰胺基相连碳上的氢,共8个;0.85(t,j=7.5hz,6h)为2
‑
乙基丁基两个甲基上的氢;1.34
–
1.27(m,7h)为2
‑
乙基丁基两个亚甲基上的氢以及与手性碳相连的一个甲基上的氢;1.45(dq,j=12.4,6.2hz,1h)为2
‑
乙基丁基上的一个次甲基上的氢。
[0027]1h
‑
nmr(600mhz,meod):δ:7.87(s,1h),7.30(t,j=7.9hz,2h),7.22
–
7.18(m,2h),7.17
–
7.13(m,1h),6.91(d,j=4.6hz,1h),6.88(d,j=4.6hz,1h),4.80(d,j=5.4hz,1h),4.43
–
4.35(m,2h),4.29(ddd,j=10.5,5.8,4.5hz,1h),4.18(t,j=5.6hz,1h),4.02(dd,j=10.9,5.8hz,1h),3.95
–
3.86(m,2h),1.45(dq,j=12.4,6.2hz,1h),1.34
–
1.27(m,7h),0.85(t,j=7.5hz,6h)。
[0028]
实施例6将1 kg瑞德西韦粗品(实施例4)加入5 kg乙醇中,搅拌加热至80
°
c溶解完全,停止加热,继续搅拌,滴加10kg纯化水,降温至10℃,搅拌6h,离心,在60℃,0.7mpa的条件下真空干燥12h,得到903g瑞德西韦精制品,收率:90.3 %,纯度:99.6%,单杂均小于0.1 %。
[0029]
实施例7将1 kg瑞德西韦粗品(实施例4)加入5 kg异丙醇中,搅拌加热至70
°
c溶解完全,停止加热,继续搅拌,滴加10kg纯化水,降温至5℃,搅拌7h,离心,在50℃,0.8mpa的条件下真空干燥10h,得到901 g瑞德西韦精制品,收率:90 .1%,纯度:99.4%,单杂均小于0.1 %。
[0030]
由实施例1
‑
4可知,本发明的三步反应均以第一溶剂为介质进行反应,仅在三步反应完成后进行后处理浓缩溶剂,减少浓缩溶剂的次数,降低工业成本;且反应条件温和、操作简单、副反应少、生产周期短、原料成本低,废液量大大减少,减轻环保压力,方便溶剂回收,大大节约了生产成本;中间体无过多后处理损失,获得产品的收率高、纯度高,适用于工
业化生产。由实施例5
‑
7可知,本发明获得产品的纯度在90 %以上,通过精制还可以进一步纯化,从而获得符合制剂要求的高纯度晶型产品。
[0031]
本具体实施例仅仅是对本发明的解释,其并不是对本发明的限制,本领域技术人员在阅读完本说明书后可以根据需要对本实施例做出没有创造性贡献的修改,但只要在本发明的权利要求范围内都受到专利法的保护。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。