1.本发明属于寡糖合成及分离技术领域,具体涉及一种光敏型氟载体及其在高效合成寡糖中的应用。
背景技术:
2.糖类化合物在许多生物过程中起着至关重要的作用,高效地获取寡糖及糖缀合物是从分子水平上深入研究糖类化合物的先决性条件。从天然产物中直接分离提取是获取糖类化合物的传统而重要的方法,但所得的寡糖结构具有微观不均一性,不利于构效关系的精准化研究。随着糖化学的发展,化学合成已经成为获得结构明确寡糖的强有力手段。然而,由于糖类化合物自身多羟基、多手性中心的特点,寡糖的高效制备面临着三个方面的挑战,即糖模块的高效制备,糖苷键的高效及高立体选择性构建,以及寡糖组装过程中的快速纯化。为了提高寡糖的合成效率,目前主要通过减少或者简化寡糖组装过程中的分离纯化过程来实现寡糖的高效制备。
3.糖的固相合成(science 2001,291,1523)主要利用一些不溶性聚合物等,通过连接臂将载体引入到糖基供体或糖基受体上,后续仅需通过洗涤即可将偶联产物与副产物及过量的反应试剂分离,该方法极大简化的分离过程,提高了寡糖的合成效率,从而使得糖的合成能够实现自动化。然而由于糖的固相合成是在非均相体系中进行的,其偶联效率很难预测。为了尽可能提高糖基化效率,往往需要在反应时使用大量的糖模块。而糖模块本身的合成工作量很大,大量糖模块的使用极大地增加了寡糖的合成成本,同时也造成了很多浪费。与此同时,糖的固相合成不能通过常规的分析方法如tlc、nmr等监测其反应过程,若需监测反应还需要将产物从载体上切断后方可按常规方法进行。
4.氟固相萃取(f-spe)技术利用高氟化的化合物与氟硅胶之间存在“氟-氟”作用力,从而只需要以憎氟性溶剂洗涤就可以除去非氟化合物,实现产物的快速分离 (tetrahedron 2006,62,11837)。pohl课题组将该技术应用于寡糖的液相自动化合成 (org.lett.2015,17,2642)。由于氟载体支载的合成是在均相体系中进行的,其糖基化效率一般不会受影响,反应也可通过常规手段来监测。尽管如此,f-spe分离技术中繁冗的转移步骤造成了化合物的损失,同时也限制了反应的规模。此外,pohl 课题组采用的氟载体不易脱除,且承载力有限。
5.基于固相合成和液相合成各自的优缺点,发明人所在课题组将固相合成和液相合成的优势结合起来,建立了一种聚四氟乙烯粒子(ptfe)协助、快速、低成本的“液相反应,固相分离”寡糖合成策略(org.lett.2020,22,2564;zl2014107046792)。在寡糖合成中,通过常规化学反应将双侧链苄基型氟载体(zl2014105380632)引入到糖上,分离纯化时,向反应混合物中加入聚四氟乙烯粒子,并通过简单的过滤洗涤就可以实现含氟化合物与非氟化合物的快速分离。基于此,通过5步糖基化反应,以48%的总收率合成了氟载体支载的复杂肿瘤相关抗原 globo-h六糖。但其中的双侧链苄基型氟载体可以与寡糖中的苄基保护基在氢解过程中一同被脱除,对于寡糖的端基快速修饰存在干扰,并影响用于后续的免疫、组
装、微阵列等研究中。
技术实现要素:
6.本发明所要解决的技术问题在于克服以往氟载体在承载力或者脱除方面的局限性,提供一类可以通过光解脱除的光敏型氟载体,以及所述氟载体在高效合成寡糖中的用途。
7.解决上述技术问题所采用的光敏型氟载体的结构式如下所示:
[0008][0009]
式中r为h或ch3。
[0010]
当r为ch3时,所述光敏型氟载体的合成方法如下:
[0011]
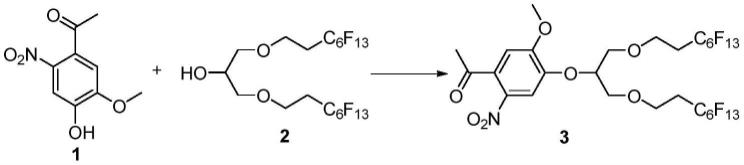
(1)将化合物2与三苯基膦、化合物1按摩尔比为1:1~2:1~2混合,并以非质子性溶剂(甲苯或者四氢呋喃)溶解,0℃到室温搅拌下,向上述溶液中加入偶氮试剂 (具体如偶氮二甲酸二乙酯、偶氮二甲酸二异丙酯等),随后于室温到150℃反应5~10小时,分离纯化产物,得到化合物3,其反应方程如下:
[0012][0013]
(2)将化合物3溶解于极性溶剂(具体如乙醇、甲醇等醇类溶剂)中,加入硼氢化钠,室温搅拌0.5~2小时,得到光敏型氟载体i,其反应方程式如下:
[0014][0015]
当r为h时,所述光敏型氟载体的合成方法如下:
[0016]
(1)将化合物4与咪唑按摩尔比为1:2溶于二氯甲烷或者四氢呋喃中,室温搅拌下加入叔丁基二甲基氯硅烷,反应1~10小时,分离纯化得到化合物5;
[0017][0018]
(2)将化合物5与三苯基膦、化合物2按摩尔比为1:1~2:1~2混合,并以非质子性溶剂(甲苯或者四氢呋喃)溶解,0℃到室温搅拌下,向所得溶液中加入偶氮试剂 (具体如偶氮二甲酸二乙酯、偶氮二甲酸二异丙酯等),随后于室温到150℃反应5~ 10小时,分离纯化产物,得到化合物6,其反应方程如下:
[0019][0020]
(2)将化合物6溶解于四氢呋喃等中,加入其摩尔量1~2倍的脱保护试剂四丁基氟化铵,室温反应2~24小时,得到光敏型氟载体ii,反应方程式为:
[0021][0022]
本发明光敏型氟载体在高效制备寡糖中的用途,具体方法如下:
[0023]
(1)将光敏型氟载体与寡糖的第一个结构单元对应的模块偶联,偶联反应完后使偶联产物和聚四氟乙烯粒子通过氟与氟之间的作用力充分吸附,过滤即得到偶联产物;然后脱除偶联产物中与下一个结构单元相连的羟基保护基,并通过聚四氟乙烯粒子纯化脱除羟基保护基的产物,使其继续偶联下一个结构单元;如此反复地进行“偶联-脱保护-偶联”操作,得到光敏型氟载体支载的寡糖;
[0024]
(2)通过光解脱除步骤(1)中所得光敏型氟载体支载的寡糖中的光敏型氟载体,得到端位具有游离羟基的寡糖,并将该寡糖通过一步化学转化成糖基供体;
[0025]
(3)采用步骤(1)的方法,反复地进行“偶联-脱保护”操作,得到脱除一个或者多个羟基保护基的光敏型氟载体支载的寡糖;
[0026]
(4)将步骤(2)所得糖基供体与步骤(3)所得寡糖偶联,得到具有更多糖单元的光敏型氟载体支载的寡糖;
[0027]
(5)将步骤(4)得到的具有更多糖单元的光敏型氟载体支载的寡糖重复上述步骤(2)至步骤(4),即实现寡糖的“汇聚式合成”,并最终获得目标寡糖。
[0028]
上述脱除光敏型氟载体的方法为:将光敏型氟载体支载的目标寡糖溶解于溶剂中,通过紫外光照射10~60分钟。其中,所用的溶剂为甲醇、乙醇、四氢呋喃、乙醚、二氯甲烷、二氧六环、乙腈等中的任意一种或两种的混合物。
[0029]
以均聚的线性寡糖为例,本发明光敏型氟载体在寡糖“汇聚式合成”中的反应过程如下所示,将光敏型氟载体与目标寡糖的第一个结构单元对应的模块a1偶联,偶联反应完后通过改变溶剂以使聚四氟乙烯粒子和光敏型氟载体支载的寡糖(以下简称氟载寡糖)通过氟与氟之间的作用力而充分吸附,从而仅通过过滤即可得到产物a2。然后,脱除产物a2中与下一个结构单元相连的基团的保护基并得到a3。随后,再加入其它结构单元对应的模块进行偶联反应,每步偶联反应完后均采用聚四氟乙烯粒子协助的寡糖分离方法来实现产物的纯化,如此重复进行“偶联-脱保护
‑ꢀ
偶联”操作,得到氟载寡糖a4。一方面对a4进行脱保护处理可以得到b1;另一方面,可以通过光解脱除光敏型氟载体,获得寡糖b2。进一步,寡糖b2可以通过一步化学反应被转化为新的糖基供体b3。通过b1与b3的偶联,可以汇聚式得到b4。 b4经脱保护后可以转化为寡糖b5。同样,寡糖b4可以被光解,并进一步转化为糖基供体c1。c1可以继续与b1等脱保护的氟载寡糖偶联得到糖单元数更多的寡糖。此外,c1也可以与
如叠氮乙醇等连接臂偶联得到化合物c2。对c2进行全脱保护处理,即可得到寡糖c3。c3可以进一步与蛋白、聚合物等高分子化合物或者其它物质偶联而用于寡糖的一些后续功能研究。
[0030][0031]
本发明的有益效果如下:
[0032]
本发明中的光敏型氟载体,不仅继承了氟载体特性,能实现寡糖的“液相反应,固相分离”;同时可以发扬“光敏”优势,易光解脱除,同时脱除条件绿色环保,避免了苛刻的催化氢解反应以及昂贵的pd催化剂的使用。此外,光敏型氟载体支载的寡糖在光解后可以转化为新的糖基供体而进行汇聚式合成及后修饰,将极大提高寡糖的制备效率,为推动寡糖的进一步研究与应用奠定基础。
具体实施方式
[0033]
下面结合实施例对本发明进一步详细说明,但本发明的保护范围不仅限于这些实施例。
[0034]
实施例1
[0035]
1、将200mg(0.26mmol)化合物2、101mg(0.38mmol)三苯基膦、75mg (0.35mmol)化合物1溶解于3ml干燥的甲苯中,冰浴冷却10分钟后,缓慢滴入60μl偶氮二甲酸二乙酯(dead),升温至110℃反应2小时后,冷却,浓缩,以乙酸乙酯与石油醚的体积比为1:3的混合液为淋洗剂硅胶柱色谱分离,得到129 mg化合物3,其产率为49%。反应方程式如下所示:
[0036][0037]
产物的结构表征数据为:1h nmr(300mhz,cdcl3)δ7.85(s,1h),6.76(s,1h), 4.66
–
4.49(m,1h),3.94(s,3h),3.87
–
3.62(m,8h),2.49(s,3h),2.54
–
2.25(m,4h)。
[0038]
2、将120mg(0.12mmol)化合物3溶解于1ml甲醇中,加入7mg(0.18mmol) 硼氢化钠,室温搅拌1小时,浓缩反应液,以乙酸乙酯与石油醚的体积比为1:2的混合液为淋洗剂进行硅胶柱色谱分离,得到106mg(0.11mmol)光敏型氟载体i,其产率为88%。反应方程式如下所示:
[0039][0040]
产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.78(s,1h),7.31(s,1h), 5.57(q,j=6.3hz,1h),4.51(p,j=4.9hz,1h),3.96(s,3h),3.86
–
3.68(m,8h),2.52
–ꢀ
2.29(m,4h),1.56(d,j=6.3hz,3h)。
[0041]
实施例2
[0042]
1、将120mg(0.60mmol)化合物4与79mg(1.16mmol)咪唑混合并以5ml 干燥的ch2cl2溶解,室温搅拌下,向所得溶液中加入117mg(0.78mmol)叔丁基二甲基氯硅烷(tbscl),反应5小时,以乙酸乙酯与石油醚的体积比为1:5的混合液为淋洗剂硅胶柱色谱分离,得到160mg(0.51mmol)化合物5,其产率为85%。反应方程式如下所示:
[0043][0044]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.77(s,1h),7.26(s,1h), 5.65(s,1h),5.08(s,2h),4.00(s,3h),0.99(s,9h),0.15(s,6h)。
[0045]
2、将86mg(0.27mmol)化合物5、143.4mg(0.18mmol)化合物2及94mg (0.36mmol)三苯基膦溶解于1ml干燥的甲苯中,冰浴冷却下,向所得溶液中滴加0.8ml含56μl偶氮二甲酸二乙酯(dead)的甲苯溶液。加完后,撤去冰浴,将反应混合物加热至100℃反应8小时后,冷却,浓缩,以乙酸乙酯与石油醚的体积比为1:10的混合液为淋洗剂硅胶柱色谱分离,得到145mg(0.13mmol)化合物 6,其产率为75%。反应方程式如下所示:
[0046][0047]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.93(s,1h),7.47(s,1h), 5.10(s,2h),4.51(p,j=4.9hz,1h),3.94(s,3h),3.85
–
3.71(m,8h),2.48
–
2.35(m, 4h),0.99(s,9h),0.15(s,6h)。
[0048]
3、将900mg(0.83mmol)化合物6溶解于5ml四氢呋喃中,加入1.4ml 1mol/l 四丁基氟化铵的四氢呋喃溶液,室温反应4小时后,tlc表明反应已经完全。向反应混合物中加入15ml乙酸乙酯,并倒入水中分液萃取,有机相以无水硫酸钠干燥,过滤,浓缩,以乙酸乙酯与石油醚的体积比为1:2的混合液为淋洗剂硅胶柱色谱分离,得到637mg(0.66mmol)光敏型氟载体ii,产率为79%。反应方程式为:
[0049][0050]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.85(s,1h),7.11(s,1h), 4.89(d,j=5.9hz,2h),4.45(p,j=4.9hz,1h),3.78
–
3.63(m,8h),2.54(t,j=6.4hz, 1h),2.39
–
2.28(m,4h)。
[0051]
实施例3
[0052]
以光敏型氟载体ii为例,对甘露糖醛酸寡糖进行了合成。具体方法如下:
[0053]
1、将47mg(0.071mmol)化合物7及43mg(0.045mmol)光敏型氟载体ii 溶解于3ml干燥的二氯甲烷中,-40℃冷却10分钟后,缓慢滴入三氟甲磺酸(tfoh) 的二氯甲烷溶液(该溶液是将5μl tfoh溶于1ml二氯甲烷得到),反应0.5小时后,tlc检测反应已经完全,加入30μl三乙胺淬灭反应。向反应液中加入807mg 聚四氟乙烯粒子后浓缩反应液,向该混合物中加入5ml体积浓度为60%的丙酮水溶液搅拌均匀,过滤,以体积浓度为60%的丙酮水溶液洗涤两次(每次5ml)后,产物以纯的丙酮解吸,旋干,得到59mg(0.041mmol)化合物8,其产率为91%。反应方程式如下所示:
[0054][0055]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.90(s,1h),7.40
–
7.35(m, 3h),7.33
–
7.24(m,8h),5.62(t,j=8.1hz,1h),5.29(d,j=15.4hz,1h),5.10(d,j= 15.4hz,1h),4.86(d,j=12.0hz,1h),4.82(d,j=12.0hz,1h),4.77(s,1h),4.58(d,j =12.5hz,1h),4.55(d,j=12.4hz,1h),4.50
–
4.46(m,1h),3.99(d,j=7.9hz,1h), 3.98
–
3.96(m,1h),3.83
–
3.72(m,8h),3.66
–
3.63(m,4h),3.61(s,3h),2.72(t,j=6.5 hz,2h),2.61
–
2.51(m,2h),2.46
–
2.35(m,4h),2.17(s,3h)。
[0056]
2、将42mg(0.029mmol)化合物8溶解于1ml二氯甲烷中,室温搅拌下,加入460μl 0.1mol/l醋酸肼的甲醇溶液,室温反应2.5小时,tlc表明反应已经完全,向反应体系中加入400mg聚四氟乙烯粒子,浓缩除去有机溶剂后,向该混合物中加入5ml体积浓度为50%的丙酮水溶液搅拌均匀,过滤,以体积浓度为50%的丙酮水溶液洗涤两次(每次5ml)后,产物以纯的丙酮解吸,旋干,得到41mg (0.029mmol)化合物9,其产率为100%。反应方程式如下所示:
[0057][0058]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.91(s,1h),7.40(d,j= 6.5hz,2h),7.35
–
7.24(m,9h),5.28(d,j=15.0hz,1h),5.11(d,j=15.0hz,1h),4.99 (d,j=12.0hz,1h),4.84(d,j=12.0hz,1h),4.68(s,1h),4.60(d,j=12.1hz,1h), 4.57(d,j=
12.0hz,1h),4.53
–
4.48(m,1h),4.32(t,j=9.7hz,1h),4.00(d,j=2.8hz, 1h),3.85
–
3.71(m,15h),3.44(dd,j=9.4,2.9hz,1h),2.93(s,1h),2.46
–
2.36(m, 4h)。
[0059]
3、将41mg(0.029mmol)化合物9及38mg(0.058mmol)化合物7溶解于 0.7ml干燥的二氯甲烷中,-40℃冷却10分钟后,缓慢滴入tfoh的二氯甲烷溶液(该溶液是将5μl tfoh溶于1ml二氯甲烷得到),反应1小时后,tlc检测反应已经完全,加入10μl三乙胺淬灭反应。向反应液中加入800mg聚四氟乙烯粒子后浓缩反应液,向该混合物中加入5ml体积浓度为60%的丙酮水溶液搅拌均匀,过滤,以体积浓度为60%的丙酮水溶液洗涤两次(每次5ml)后,产物以纯的丙酮解吸,旋干,得到50.6mg(0.028mmol)化合物10,其产率为97%。反应方程式如下所示:
[0060][0061]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.92
–
7.89(m,1h),7.37 (d,j=7.1hz,2h),7.35
–
7.30(m,5h),7.29
–
7.20(m,14h),5.46(t,j=9.6hz,1h), 5.36(d,j=15.6hz,1h),5.06(d,j=15.6hz,1h),4.83
–
4.77(m,2h),4.76
–
4.72(m, 2h),4.65(d,j=10.2hz,2h),4.54
–
4.49(m,2h),4.49
–
4.45(m,1h),4.44(d,j=12.4 hz,1h),4.03(d,j=6.7hz,1h),3.96
–
3.93(m,1h),3.86(dd,j=7.3,2.9hz,1h),3.83 (d,j=2.7hz,1h),3.81
–
3.68(m,9h),3.64(d,j=4.7hz,1h),3.62(d,j=3.3hz,1h), 3.58(s,3h),3.53(t,j=3.8hz,6h),3.46(dd,j=9.6,2.7hz,1h),2.69(t,j=6.8hz, 2h),2.59
–
2.49(m,2h),2.45
–
2.35(m,4h),2.16(s,3h)。
[0062]
4、将50mg(0.028mmol)化合物10溶解于2.8ml四氢呋喃中,以365nm的紫外灯照射10分钟后,tlc检测反应已经完全。浓缩,以乙酸乙酯与石油醚的体积比为1:2的混合液为淋洗剂硅胶柱色谱分离,得到20mg(0.024mmol)化合物 11,其产率为86%。反应方程式如下所示:
[0063][0064]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.33
–
7.13(m,20h),5.48
ꢀ–
5.41(m,2h),4.75(d,j=12.4hz,1h),4.66(d,j=12.4hz,1h),4.57
–
4.52(m,3h), 4.48(d,j=12.0hz,1h),4.44(d,j=12.4hz,1h),4.41(dd,j=4.9,3.6hz,1h),4.36(d, j=3.4hz,1h),4.34(d,j=12.3hz,1h),4.13(dd,j=4.7,2.7hz,1h),3.82(d,j=2.5 hz,1h),3.78(d,j=9.3hz,1h),3.59
–
3.51(m,5h),3.47(s,3h),3.42(dd,j=9.5,2.8 hz,1h),3.09(d,j=4.7hz,1h),2.68
–
2.60(m,2h),2.55
–
2.41(m,2h),2.10(s,3h)。
[0065]
5、将242mg(0.28mmol)化合物11及112mg(0.54mmol)n-苯基三氟乙酰氯溶解于3ml丙酮中,室温搅拌下,加入77mg(0.56mmol)碳酸钾,反应2小时后,tlc检测反应已经完全,浓缩,以乙酸乙酯与石油醚的体积比为1:2混合液 (其中添加混合液体积5%的三乙胺)为淋洗剂硅胶柱色谱分离,得280mg(0.27 mmol)化合物12,其产率为97%。反应方程式如下所
示:
[0066][0067]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.38(d,j=7.0hz,2h), 7.36
–
7.30(m,3h),7.30
–
7.20(m,17h),7.09(t,j=7.5hz,1h),6.82(d,j=7.5hz, 2h),6.47(s,1h),5.51(t,j=9.5hz,1h),4.81(d,j=12.3hz,1h),4.76(d,j=12.4hz, 1h),4.69(d,j=11.9hz,1h),4.64(s,1h),4.59(d,j=12.0hz,1h),4.57
–
4.52(m,3h), 4.46(t,j=9.4hz,2h),4.36(d,j=3.8hz,1h),4.17(s,1h),3.87(d,j=2.4hz,1h), 3.86
–
3.79(d,j=9.4hz,2h),3.64(s,3h),3.56(s,3h),3.49(dd,j=9.5,2.8hz,1h), 2.72(t,j=6.7hz,2h),2.63
–
2.48(m,2h),2.17(s,3h)。
[0068]
6、将116mg(0.064mmol)化合物10溶解于2ml二氯甲烷中,室温搅拌下,加入0.26ml 0.5mol/l醋酸肼的甲醇溶液,室温反应2小时,tlc表明反应已经完全,向反应体系中加入1g聚四氟乙烯粒子,浓缩除去有机溶剂后,向该混合物中加入5ml体积浓度为50%的丙酮水溶液搅拌均匀,过滤,以体积浓度为50%的丙酮水溶液洗涤两次(每次5ml)后,产物以纯的丙酮解吸,旋干,得到112mg(0.065 mmol)化合物12,其产率为100%。反应方程式如下所示:
[0069][0070]
产物的结构表征数据为:1h nmr(400mhz,cdcl3)δ7.90(s,1h),7.40
–
7.19(m, 21h),5.36(d,j=15.7hz,1h),5.06(d,j=15.6hz,1h),4.85
–
4.79(m,3h),4.75
–
4.68(m,3h),4.67
–
4.62(m,2h),4.59
–
4.55(m,3h),4.49
–
4.45(m,1h),4.23(t,j= 9.5hz,1h),4.07(d,j=6.5hz,1h),3.96(s,1h),3.89(dd,j=7.1,2.9hz,1h),3.83(d, j=2.4hz,1h),3.82
–
3.69(m,9h),3.64(s,3h),3.56(d,j=9.1hz,1h),3.54(s,3h), 3.52(s,3h),3.33(dd,j=9.4,2.7hz,1h),2.96(s,1h),2.50
–
2.30(m,4h)。
[0071]
7、将112mg(0.066mmol)化合物13及186mg(0.18mmol)化合物12溶解于1ml干燥的二氯甲烷中,-40℃冷却10分钟后,缓慢滴入tfoh的二氯甲烷溶液(该溶液是将10μl tfoh溶于1ml二氯甲烷得到,取100μl),反应1小时后, tlc检测反应已经完全,加入20μl三乙胺淬灭反应。向反应液中加入3g聚四氟乙烯粒子后浓缩反应液,向该混合物中加入10ml体积浓度为65%的丙酮水溶液搅拌均匀,过滤,以体积浓度为65%的丙酮水溶液洗涤两次(每次10ml)后,再以 10ml体积浓度为70%的丙酮水溶液洗涤洗涤一次,产物以纯的丙酮解吸,旋干,得到170mg(0.066mmol)化合物14,其产率为100%。反应方程式如下所示:
[0072][0073]
产物的结构表征数据为:1h nmr(600mhz,cdcl3)δ7.90(s,1h),7.37
–
7.18(m, 41h),5.40(t,j=9.8hz,1h),5.35(d,j=15.5hz,1h),5.04(d,j=15.5hz,1h),4.83
–ꢀ
4.75
(m,4h),4.74
–
4.67(m,6h),4.66
–
4.58(m,4h),4.57
–
4.51(m,3h),4.50
–
4.45 (m,2h),4.40(d,j=12.4hz,2h),4.36(t,j=9.0hz,1h),4.29(t,j=9.3hz,1h),4.02 (d,j=6.6hz,1h),3.92(s,1h),3.84
–
3.70(m,12h),3.67
–
3.63(m,2h),3.61
–
3.57(m, 1h),3.56
–
3.52(m,3h),3.52(s,3h),3.51(s,3h),3.50(s,3h),3.43(s,3h),3.40(dd,j =9.7,2.8hz,1h),2.69
–
2.65(m,2h),2.57
–
2.47(m,2h),2.46
–
2.35(m,4h),2.15(s, 3h)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
