一种ep2受体选择性的前列腺素e2激动剂的制备方法
技术领域
1.本发明属于药物化学合成技术领域,具体涉及一种ep2受体选择性的前列腺素e2激动剂的制备方法。
背景技术:
2.ep2受体选择性的前列腺素e2激动剂简称evatanepag,是一种ep4拮抗剂,ep4拮抗剂是一种阻断通过pge2与前列腺素e受体4(ep4)的相互作用的前列腺素e2(pge2)信号传导。
3.目前并没有相应的文献公开本技术的evatanepag的制备方法。
技术实现要素:
4.针对现有技术中存在的不足和限制,本发明的目的是提供一种ep2受体选择性的前列腺素e2激动剂的制备方法,以克服现有技术的不足。
5.本发明的目的是这样来达到的,一种ep2受体选择性的前列腺素e2激动剂的制备方法,包括如下步骤:
6.s1:制备化合物im-1:
7.向单口瓶中加入化合物1和碳酸钾及有机溶剂,在搅拌条件下向单口瓶中再次加入化合物2,并通过油浴加热,反应结束后将至室温加入乙酸乙酯和水,并用乙酸乙酯萃取水相,合并有机相,并将有机相用饱和食盐水洗涤后用无水硫酸钠干燥,过滤,滤液旋干过柱得到化合物im-1,反应式如下:
[0008][0009]
s2:制备化合物im-2:
[0010]
将化合物im-1和化合物4溶于有机溶剂,加入催化剂硼氢化钠并搅拌,反应结束后将反应液倒入冰水中,然后用二氯甲烷萃取三遍,合并二氯甲烷旋干过柱得到化合物im-2,反应式如下:
[0011][0012]
s3:制备化合物im-3:
[0013]
将化合物im-2溶于有机溶剂,加入三乙胺,然后加入化合物6并搅拌一段时间后,
恢复至室温继续搅拌,反应结束后向反应液加碳酸氢钠水溶液淬灭,加水,然后用二氯甲烷萃取,合并二氯甲烷旋干过柱得到化合物im-3,反应式如下:
[0014][0015]
s4:制备目标产物:
[0016]
将化合物im-3溶于有机溶剂,加入三氟乙酸搅拌一段时间后,升到室温继续搅拌,反应结束后反应液旋干后溶于乙酸乙酯中,加水并调节ph到5,析出大量固体,过滤得到滤饼和滤液,滤液用乙酸乙酯萃取两遍,合并乙酸乙酯旋干过柱,滤饼也过柱,过柱后的产品用乙醇重结晶得到目标产物,反应式如下:
[0017][0018]
进一步的:步骤s1中化合物1和化合物2的质量比为(3.5-4.5):(8-10),化合物1和碳酸钾的质量比为(3.5-4.5):(100-150)。
[0019]
进一步的:步骤s1中有机溶剂为dmf,且加入化合物2后油浴加热至100度;步骤s1中的过滤柱为pe和ea两种溶剂混合的过滤柱,且ea/pe=0~10%。
[0020]
进一步的:步骤s2中有机溶剂为甲醇,且化合物im-1和化合物4溶于有机溶剂后在室温下搅拌2小时,然后冰浴冷却至0度后加入催化剂硼氢化钠,加入催化剂硼氢化钠后在0度条件下搅拌1小时;步骤s2中的过滤柱为pe和ea两种溶剂混合的过滤柱,且ea/pe=0~20%。
[0021]
进一步的:步骤s2中化合物im-1和化合物4的质量比为(35-45):(25-35)。
[0022]
进一步的:步骤s3中有机溶剂为二氯甲烷,且加入二氯甲烷后冰浴冷却到0度后加入三乙胺;加入化合物6后需要0度搅拌30分钟,恢复至室温继续搅拌2小时。
[0023]
进一步的:步骤s3中的过滤柱为pe和ea两种溶剂混合的过滤柱,且ea/pe=0~35%。
[0024]
进一步的:步骤s4中有机溶剂为二氯甲烷,且加入二氯甲烷后冰浴冷却到0度后加入加入三氟乙酸;加入加入三氟乙酸后需要0度搅拌10分钟,恢复至室温继续搅拌2小时。
[0025]
进一步的:步骤s4中调节ph采用的是碳酸氢钠水溶液。
[0026]
进一步的:步骤s3中的过滤柱为ch3oh和dcm两种溶剂混合的过滤柱,且ch3oh/dcm=0~5%。
[0027]
本发明具有如下有益效果:
[0028]
本发明的制备方法比较温和,不会发生爆炸等,从而可以避免制备过程中出现爆炸等生产事故,另外工艺操作简单,具有便于操作的优点;
[0029]
本发明工艺路线简洁,产生的杂质较少,且制备过程中无污染物产生,体现了绿色环保的优点;
[0030]
本发明起始原料和所用的试剂易得,因此可以满足大量制备的需求,能够实现工业化生产。
附图说明
[0031]
图1是对化合物im-1进行核磁共振氢谱分析的结果;
[0032]
图2是对化合物im-1进行色谱分析的结果;
[0033]
图3是对化合物im-2进行核磁共振氢谱分析的结果;
[0034]
图4是对化合物im-2进行色谱分析的结果;
[0035]
图5是对化合物im-3进行核磁共振氢谱分析的结果;
[0036]
图6是对化合物im-3进行色谱分析的结果;
[0037]
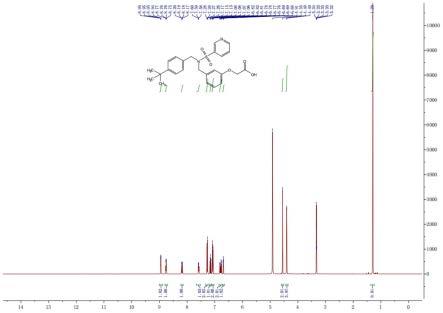
图7是对目标产物evatanepag进行核磁共振氢谱分析的结果;
[0038]
图8是对目标产物evatanepag进行色谱分析的结果。
具体实施方式
[0039]
以下结合实施例对本发明技术方案作进一步非限制性的详细说明。
[0040]
实施例1
[0041]
1)制备化合物im-1:
[0042]
1升的单口瓶里加入40克化合物1,135克碳酸钾,加入500毫升dmf,搅拌下加入90克化合物2,油浴加热到100度反应4小时,lcms显示反应结束,降至室温,加入乙酸乙酯和水,水相用乙酸乙酯萃取两遍,合并有机相,用饱和食盐水洗涤两次,有机相用无水硫酸钠干燥,过滤,滤液旋干过柱(ea/pe=0~10%),得到64.2克化合物im-1收率83%,反应式如下:
[0043][0044]
对得到的化合物im-1进行核磁共振氢谱分析和色谱分析,核磁共振氢谱分析的结果如图1所示,色谱分析的结果如图2所示;
[0045]
2)制备化合物im-2:
[0046]
40克化合物im-1和30克化合物4溶于400毫升甲醇里,室温搅拌2小时,冰浴冷却到0度,慢慢加入9.6克硼氢化钠下,加完后0度搅拌1小时,lcms显示反应结束,反应液倒入冰水中,然后用二氯甲烷萃取三遍,合并二氯甲烷旋干过柱(ea/pe=0~20%)得到54.5克化合物im-2,收率85%,反应式如下:
[0047]
[0048]
对得到的化合物im-2进行核磁共振氢谱分析和色谱分析,核磁共振氢谱分析的结果如图3所示,色谱分析的结果如图4所示;
[0049]
3)制备化合物im-3:
[0050]
46克化合物im-2溶于400毫升二氯甲烷里,冰浴冷却到0度,加入37毫升三乙胺,慢慢加入23.5克化合物6,加完后0度搅拌30分钟,升到室温搅拌2小时,lcms显示反应结束,反应液加碳酸氢钠水溶液淬灭,加水,然后用二氯甲烷萃取三遍,合并二氯甲烷旋干过柱(ea/pe=0~35%)得到52.8克化合物im-3,收率84%,反应式如下:
[0051]
对得到的化合物im-3进行核磁共振氢谱分析和色谱分析,核磁共振氢谱分析的结果如图5所示,色谱分析的结果如图6所示;
[0052]
4)制备目标产物evatanepag:
[0053]
4.2克化合im-3溶于50毫升二氯甲烷里,冰浴冷却到0度,加入8毫升三氟乙酸,加完后0度搅拌10分钟,升到室温搅拌2小时,lcms显示反应结束,反应液旋干后溶于乙酸乙酯中,加水,用碳酸氢钠水溶液调节ph到5左右,析出大量固体,过滤,滤饼为粗产品,滤液用乙酸乙酯萃取两遍,合并乙酸乙酯旋干过柱(ch3oh/dcm=0~5%),滤饼也过柱(ch3oh/dcm=0~5%),得到的产品用乙醇重结晶得到2.8克白色粉末状固体evatanepag,收率74%,该反应的反应式如下:
[0054][0055]
对得到的目标产物evatanepag进行核磁共振氢谱分析和色谱分析,核磁共振氢谱分析的结果如图7所示,色谱分析的结果如图8所示。
[0056]
以上结合附图详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种等同变换,这些等同变换均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。