1.本发明涉及医药技术领域,尤其涉及一种双酮类化合物的制备方法和咪唑类衍生物的制备方法。
背景技术:
2.咪唑基是生物体内组氨酸、核糖核酸(rna)和脱氧核糖核酸(dna)嘌呤的组成部分。咪唑环中的氢原子可以在两个氮原子之间迁移,具有良好的电子转移性和易功能化的特点,咪唑类衍生物中具有广泛生物活性。示例性地,专利申请cn103025731a公开了如下化学式i所示的化合物,该化合物具有抑制alk5和/或alk4的生物活性,可以用于抑制肿瘤及异常增殖性疾病,具有成为新型抗肿瘤药物的前景。
[0003][0004]
其中r
10
为碳原子数为1~6的烷基等基团;r
20
为f、cl、br、碳原子数为1~6的烷基、碳原子数为2~6的烯基、碳原子数为2~6的炔基、碳原子数为1~6的烷氧基等基团。
[0005]
化学式ii所示的双酮类化合物,为合成化学式i所示的咪唑类衍生物的关键中间体。
[0006][0007]
现有技术中,化学式ii所示的双酮类化合物采用如下合成路线1所示的方法进行合成:
[0008][0009]
根据如上合成步骤,在制备中间体a-ii时,需要将原料a-i与苯胺和二苯基亚磷酸
酯在氯化氧锆作用下,得到n-p缩醛中间体b-ii;该步骤中需要使用对人体高危害的氯化氧锆,且需要使用异丙醇。这不仅会对水体产生严重的污染,而且异丙醇容易残留而导致最终产品不符合ich(international conference on harmonization of technical requirements for registration of pharmaceuticals for human use,人用药物注册技术要求国际协调会议)对异丙醇的规定(异丙醇残留量不超过0.5%)。在制备中间体c-iii时,需要将中间体b-ii与[1,2,4]三氮唑[1,5-a]吡啶-6-甲醛在碱性条件下进一步偶联,酸性水解生成一元酮,得到中间体c-iii。在该步骤中需要使用大量的碳酸铯并采用四氢呋喃和异丙醇做溶剂,在后处理过程中需要反复调节酸碱,不仅工序复杂而且在大规模生产中容易损坏设备,产生的废水量大,且异丙醇容易残留而导致最终产品不符合ich对异丙醇的规定。在制备化学式2所示的双酮类化合物的过程中,采用dmso(二甲基亚砜)和hbr对中间体c-iii进行氧化,在反应中应用了大量的hbr,不仅对设备的损害大,而且废水处理量大,对环境污染严重。
[0010]
所述背景技术部分公开的上述信息仅用于加强对本发明的背景的理解,因此它可以包括不构成对本领域普通技术人员已知的现有技术的信息。
技术实现要素:
[0011]
本发明的目的在于提供一种双酮类化合物的制备方法和咪唑类衍生物的制备方法,降低咪唑类衍生物在制备过程中的环境污染。
[0012]
为实现上述发明目的,本发明采用如下技术方案:
[0013]
根据本发明的第一个方面,提供一种双酮类化合物的制备方法,所述双酮类化合物的结构式如化学式2所示:
[0014][0015]
其中,各r1独立地选自碳原子数为氘、卤素基团、氰基、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基或碳原子数为1~6的卤代烷基;
[0016]
所述双酮类化合物的制备方法包括:
[0017]
步骤一、
[0018][0019]
化学式p1所示的化合物、硫和溴乙烷反应,生成化学式p2所示的化合物;
[0020]
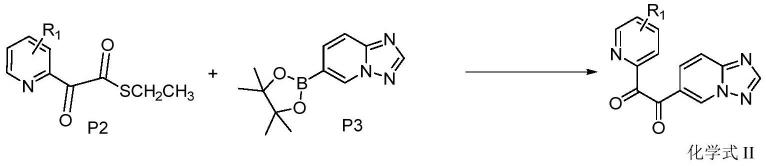
步骤二、
pr、-ch2ch2ch3),异丙基(i-pr、-ch(ch3)2),正丁基(n-bu、-ch2ch2ch2ch3),异丁基(i-bu、-ch2ch(ch3)2),仲丁基(s-bu、-ch(ch3)ch2ch3),叔丁基(t-bu、-c(ch3)3)等。
[0033]
在本技术中,作为取代基的卤素基团,有氟、氯、溴或碘。
[0034]
在本技术中,“烷氧基”表示烷基基团通过氧原子与分子其余部分相连,其中烷基基团具有如本发明所述的含义。在一实施方案中,烷氧基基团含有1-6个碳原子;在另一实施方案中,烷氧基基团含有1-4个碳原子;在又一实施方案中,烷氧基基团含有1-3个碳原子。所述烷氧基基团可以任选地被一个或多个本发明描述的取代基所取代。烷氧基基团的实例包括,但并不限于,甲氧基(meo、-och3),乙氧基(eto、-och2ch3),1-丙氧基(n-pro、n-丙氧基、-och2ch2ch3),2-丙氧基(i-pro、i-丙氧基、-och(ch3)2),1-丁氧基(n-buo、n-丁氧基、-och2ch2ch2ch3),2-甲基-l-丙氧基(i-buo、i-丁氧基、-och2ch(ch3)2),2-丁氧基(s-buo、s-丁氧基、-och(ch3)ch2ch3),2-甲基-2-丙氧基(t-buo、t-丁氧基、-oc(ch3)3),等等。
[0035]
在本技术中,“卤代烷基”或“卤代烷氧基”表示烷基或烷氧基基团被一个或多个卤素原子所取代,其中烷基和烷氧基基团具有如本发明所述的含义,这样的实例包含,但并不限于,三氟甲基、三氟甲氧基等。在一实施方案中,c
1-c6卤代烷基包含氟取代的c
1-c6烷基;在另一实施方案中,c
1-c4卤代烷基包含氟取代的c
1-c4烷基;在又一实施方案中,c
1-c2卤代烷基包含氟取代的c
1-c2烷基。
[0036]
本发明提供一种双酮类化合物的制备方法,所述双酮类化合物的结构式如化学式ii所示:
[0037][0038]
其中,r1选自碳原子数为氘、卤素基团、氰基、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基或碳原子数为1~6的卤代烷基。
[0039]
可选地,r1选自甲基、乙基、异丙基、叔丁基。
[0040]
在本技术中,化学式2所示的双酮类化合物选自如下所示结构:
[0041][0042]
如化合物2所示的双酮类化合物的制备方法包括以下步骤:
[0043]
步骤一、
[0044][0045]
化学式p1所示的化合物、硫单质和溴乙烷反应,生成化学式p2所示的化合物;
[0046]
步骤二、
[0047][0048]
化学式p2所示的化合物和化学式p3所示的化合物发生偶联反应,生成化学式ii所示的双酮类化合物。
[0049]
根据本发明提供的双酮类化合物的制备方法,在制备化学式ii所示的双酮类化合物时,无需采用氯化氧锆等对人有严重危害的化学品,能够降低这些危险品对人体和环境的危害。不仅如此,在制备过程中避免大量应用酸,避免产生大量酸性废水进而减少废水处理量并降低环境污染。
[0050]
可选地,在步骤一中,化学式p1所示的化合物、硫单质、溴乙烷、第一碱和第一溶剂的反应混合物进行反应,生成化学式p2所示的化合物。如此,在步骤一中,避免采用大量酸性溶液,而是在碱性环境中反应,避免了大量酸性废液可能产生的污染。不仅如此,由于在碱性条件下反应,因此反应产物不会成盐,在反应结束后可以无需调节ph而直接通过萃取等方法分离产物,减少了后处理工序和减少了废液量,能够简化后处理过程。
[0051]
进一步可选地,在步骤一中,硫单质为环八硫(s8)。
[0052]
进一步可选地,在步骤一中,按照摩尔量计,化学式p1所示的化合物:硫单质=1:(1~2)。可选地,按照摩尔量计,化学式p1所示的化合物:硫单质=1:(1.2~2)。可选地,按照摩尔量计,化学式p1所示的化合物:硫单质=1:(1.5~1.8)。
[0053]
进一步可选地,在步骤一中,按照摩尔量计,化学式p1所示的化合物:溴乙烷=1:(1.2~2)。可选地,按照摩尔量计,化学式p6所示的化合物:溴乙烷=1:(1.2~1.8)。可选地,按照摩尔量计,化学式p6所示的化合物:溴乙烷=1:1.8。
[0054]
进一步可选地,在步骤一中,第一碱为无机碱或者有机弱酸的碱金属盐,例如可以为碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠、乙酸钾、氢氧化钠等常用的无机碱或者有机弱酸的碱金属盐。一方面,这可以避免采用有机碱,例如采用三乙胺、二异丙基乙胺、吡啶等有机碱,降低废水中的有机物含量,减少环境污染。另一方面,无机碱或者有机弱酸的碱金属盐不仅对环境的污染本身小于有机碱,而且还可以通过浓缩的方式生成并回收固体废物而降低对环境的污染。不仅如此,这些无机碱或者有机弱酸的碱金属盐在水相中的溶解度更好,可以在萃取过程中与反应产物有效分离,降低了反应产物的纯化难度。
[0055]
可选地,在步骤一中,第一碱选自碳酸钾、碳酸钠、碳酸氢钠、碳酸氢钾、乙酸钾、氢氧化钠中的一种或者多种,以保证第一碱具有高的溶解度和低廉的成本。更进一步可选地,第一碱选自碳酸氢钠、碳酸氢钾或乙酸钾中的一种。
[0056]
进一步可选地,在步骤一中,按照摩尔量计,化学式p1所示的化合物:第一碱=1:(1~2)。可选地,按照摩尔量计,化学式p1所示的化合物:第一碱=1:(1.6~2.0)。进一步可选地,按照摩尔量计,化学式p1所示的化合物:第一碱=1:2.0。
[0057]
进一步可选地,在步骤一中,第一碱的摩尔量不小于溴乙烷的摩尔量。
[0058]
进一步可选地,在步骤一中,所述第一溶剂为第一有机溶剂和水的混合物,其中,所述第一有机溶剂包括芳香烃类溶剂、酰胺类溶剂、醚类溶剂、腈类溶剂中的一种或者多种;其中,按照体积计,所述第一有机溶剂:水=1:(0.2~0.5)。如此,在该步骤一中,可以采用有机溶剂和水的混合溶剂,在反应结束后可以通过萃取分离产品。
[0059]
更具体地,在步骤一中,芳香烃类溶剂包括但不限于甲苯、二甲苯、均三甲苯等。酰胺类溶剂包括但不限于二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮等。醚类溶剂包括但不限于乙醚、甲基叔丁基醚、环戊基甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、苯甲醚等。腈类溶剂包括但不限于乙腈、戊腈等。
[0060]
可选地,在步骤一中,第一有机溶剂选自甲苯、二甲基甲酰胺、环戊基甲醚、二氧六环、苯甲醚、乙腈中的一种。进一步可选地,所述第一有机溶剂为甲苯或苯甲醚,以提高反应效率。
[0061]
可选地,在步骤一中,按照体积计,所述第一有机溶剂:水=1:0.2。
[0062]
可选地,在步骤一中,化学式p1所示的化合物的摩尔量:第一溶剂的体积=1mmol:(2~3)ml。
[0063]
进一步可选的,在步骤一中,制备化学式p2所示的化合物的反应体系中还包括第一相转移催化剂,以便提高反应效率。即,化学式p1所示的化合物、硫单质、溴乙烷、第一碱、第一相转移催化剂和第一溶剂的混合物进行反应,生成化学式p2所示的化合物。
[0064]
可选地,在步骤一中,所述反应混合物中还包括第一相转移催化剂,所述第一相转移催化剂选自四丁基溴化铵、18-冠-6-醚、15-冠-5-醚、tebac(苄基三乙基氯化铵)、tbac(四丁基氯化铵)。进一步可选地,第一相转移催化剂为四丁基溴化铵。
[0065]
可选地,在步骤一中,按照摩尔量计,化学式p1所示的化合物:第一相转移催化剂=1:(0.05~0.2)。进一步可选地,按照摩尔量计,化学式p1所示的化合物:第一相转移催化剂=1:0.1。
[0066]
进一步可选地,在步骤一中,包含有化学式p1所示的化合物、硫单质、溴乙烷、第一碱和第一溶剂的混合物在60℃~85℃下进行反应,即反应温度为60℃~85℃。可选地,反应温度为70℃~85℃。可选地,反应温度为80℃~85℃。
[0067]
在本发明的一种更具体的实施方式中,步骤一中,反应体系为化学式p1所示的化合物、硫单质、溴乙烷、第一碱、第一相转移催化剂和第一溶剂的混合物,其中,按照摩尔量计,化学式p6所示的化合物:硫单质:溴乙烷:第一碱:第一相转移催化剂=1:1.5:1.8:2:0.1;第一溶剂包括甲苯和水,其中,甲苯的体积:水的体积=10:2。
[0068]
可选地,在步骤二中,化学式p2所示的化合物、化学式p3所示的化合物、钯催化剂、第二碱和第二溶剂的混合物反应,生成化学式2所示的双酮类化合物。在步骤二中,该合成路线相比于现有技术中的制备方式而言,避免采用大量酸性溶液,而是在碱性环境中反应,避免了大量酸性废液可能产生的污染。不仅如此,由于在碱性条件下反应,因此反应产物不会成盐,在反应结束后可以无需调节ph而直接通过萃取等方法分离产物,减少了后处理工
序和减少了废液量,能够简化后处理过程。
[0069]
进一步可选地,在步骤二中,按照摩尔量计,化学式p2所示的化合物:化学式p3所示的化合物=1:(1~1.2)。
[0070]
进一步可选地,在步骤二中,钯催化剂选自醋酸钯、氯化钯、四(三苯基膦)钯、二氯二叔丁基-(4-二甲基氨基苯基)磷钯(ii)或者其他二价钯催化剂。可选地,钯催化剂为二氯二叔丁基-(4-二甲基氨基苯基)磷钯(ii)(pd132)。
[0071]
进一步可选地,在步骤二中,按照摩尔量计,化学式p2所示的化合物:钯催化剂=1:(0.005~0.01)。可选地,按照摩尔量计,化学式p2所示的化合物:钯催化剂=1:0.005。
[0072]
进一步可选地,在步骤二中,第二碱为无机碱或者有机弱酸的碱金属盐,例如碳酸钾、碳酸钠、碳酸氢钾、碳酸氢钠、氢氧化钠、乙酸钾等常用的无机碱或者有机弱酸的碱金属盐。一方面,这可以避免采用有机碱,例如采用三乙胺、二异丙基乙胺、吡啶等有机碱,降低废水中的有机物含量,减少环境污染。另一方面,无机碱或者有机弱酸的碱金属盐不仅对环境的污染本身小于有机碱,而且还可以通过浓缩的方式生成并回收固体废物而降低对环境的污染。不仅如此,这些无机碱或者有机弱酸的碱金属盐在水相中的溶解度更好,可以在萃取过程中与反应产物有效分离,降低了反应产物的纯化难度。
[0073]
可选地,第二碱选自碳酸钾、碳酸钠、碳酸氢钠、碳酸氢钾、乙酸钾、氢氧化钠中的一种或者多种,以保证第二碱具有高的溶解度和低廉的成本。更进一步可选地,第二碱选自碳酸钠、乙酸钾或碳酸钾中的一种。
[0074]
进一步可选地,按照摩尔量计,化学式p2所示的化合物:第二碱=1:(1~2)。可选地,按照摩尔量计,化学式p2所示的化合物:第二碱=1:(1.5~2.0)。进一步可选地,按照摩尔量计,化学式p2所示的化合物:第二碱=1:2.0。
[0075]
进一步可选地,所述第二溶剂为第二有机溶剂和水的混合物,其中,所述第二有机溶剂包括醇类溶剂、芳香烃类溶剂、酰胺类溶剂、醚类溶剂、腈类溶剂中的一种或者多种;其中,按照体积计,所述第二有机溶剂:水=1:(0.2~0.5)。如此,在该步骤二中,优选有机溶剂和水的混合溶剂,在反应结束后可以直接萃取分离产品。
[0076]
其中,醇类溶剂包括但不限于甲醇、乙醇、乙二醇、叔丁醇等c1-c4的低级醇。芳香烃类溶剂包括但不限于甲苯、二甲苯、均三甲苯等。酰胺类溶剂包括但不限于二甲基甲酰胺、二甲基乙酰胺、n-甲基吡咯烷酮等。醚类溶剂包括但不限于乙醚、甲基叔丁基醚、环戊基甲醚、1,4-二氧六环、四氢呋喃、2-甲基四氢呋喃、乙二醇二甲醚、苯甲醚等。腈类溶剂包括但不限于乙腈、戊腈等。
[0077]
可选地,第二有机溶剂选自醇类溶剂、甲苯、二甲基甲酰胺、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、乙腈中的一种或者多种的混合溶剂。进一步可选地,所述第二有机溶剂为甲苯,以提高反应效率。
[0078]
可选地,按照体积计,所述第二有机溶剂:水=3:1。
[0079]
可选地,化学式p2所示的化合物的摩尔量:第二溶剂的体积=1mmol:(3~5)ml。
[0080]
进一步可选地,在步骤二中,所述混合物中还包括第二相转移催化剂。即,化学式p2所示的化合物、化学式p3所示的化合物、钯催化剂、第二碱、第二相转移催化剂和第二溶剂的混合物反应,生成化学式2所示的双酮类化合物。
[0081]
可选地,第二相转移催化剂选自四丁基溴化铵、18-冠-6-醚、15-冠-5-醚、tebac
(苄基三乙基氯化铵)、tbac(四丁基氯化铵)。进一步可选地,第二相转移催化剂为四丁基溴化铵。
[0082]
可选地,按照摩尔量计,化学式p2所示的化合物:第二相转移催化剂=1:(0.05~0.1)。进一步可选地,按照摩尔量计,化学式p2所示的化合物:第二相转移催化剂=1:0.05。
[0083]
进一步可选地,包含有化学式p2所示的化合物、化学式p3所示的化合物、钯催化剂、第二碱和第二溶剂的混合物在60℃~80℃下进行反应,即反应温度为60℃~80℃。可选地,反应为度为65℃~75℃。可选地,反应为度为65℃~70℃。
[0084]
在本发明的一种实施方式中,反应体系为化学式p2所示的化合物、化学式p3所示的化合物、钯催化剂、第二碱、第二相转移催化剂和第二溶剂的混合物。其中,按照摩尔量计,化学式p2所示的化合物:化学式p3所示的化合物:钯催化剂:第二碱:第二相转移催化剂=1:1.05:0.005:2:0.05。第二溶剂包括甲苯和水,其中,甲苯的体积:水的体积=3:1。
[0085]
本发明还提供一种咪唑类衍生物的制备方法。所述咪唑类衍生物的结构如化学式1所示:
[0086][0087]
其中,r1独立地选自氘、卤素基团、氰基、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基或碳原子数为1~6的卤代烷基;r2为氟、氯、溴、碳原子数为1~6的烷基、碳原子数为1~6的烷氧基或碳原子数为1~6的卤代烷基;
[0088]
可选地,化学式1中r1选自氟、氯、甲基、乙基、异丙基或叔丁基。
[0089]
可选地,所述咪唑类衍生物选自如下所示的化合物:
[0090][0091]
本技术所述的咪唑衍生物的制备方法包括上述的双酮类化合物的制备方法。因此,该咪唑衍生物的制备方法,具有与上述的双酮类化合物的制备方法相同的有益效果,本发明在此不再赘述。
[0092]
可选地,所述咪唑类衍生物的制备方法还包括:
[0093]
步骤三、
[0094][0095]
化学式ii所示的化合物、乙二醛二甲缩醛和铵盐反应,生成化学式p4所示的化合物;
[0096]
步骤四:
[0097][0098]
化学式p4所示的化合物生成化学式p5所示的化合物;
[0099]
步骤五:
[0100][0101]
化学式p5所示的化合物和化学式p6所示的化合物反应,生成化学式1所示的化合物。
[0102]
可选地,在步骤三中,按照摩尔量计,化学式ii所示的化合物:乙二醛二甲缩醛=1:(2~2.5)。可选地,按照摩尔量计,化学式ii所示的化合物:乙二醛二甲缩醛=1:2.0。
[0103]
可选地,在步骤三中,按照摩尔量计,化学式ii所示的化合物:铵盐=1:(2~2.5)。可选地,按照摩尔量计,化学式2所示的化合物:铵盐=1:2.2。
[0104]
可选地,在步骤三中,铵盐选自醋酸铵、甲酸铵、氯化铵、碳酸氢铵或者其他铵盐。
[0105]
在本发明的一种实施方式中,化学式ii所示的化合物、乙二醛二甲缩醛、铵盐和第三溶剂的混合物反应,生成化学式p4所示的化合物。其中,乙二醛二甲缩醛加入温度为-10℃~10℃。可选地,加入温度为-5℃~5℃,可选地,加入温度为-5℃~0℃。
[0106]
在本发明的另一种实施方式中,在步骤三中,可以在第一反应温度下,向化学式ii所示的化合物与第三溶剂形成的反应溶液中加入乙二醛二甲缩醛,并在第一反应温度下反应至完全;然后再向反应体系中加入铵盐,然后在第二反应温度下反应至完全,生成化学式p4所示的化合物。
[0107]
可选地,第一反应温度为-10℃~10℃。进一步可选地,第一反应温度为-5℃~5℃。
[0108]
在一种进一步的方案中,第二反应温度为环境温度。
[0109]
在另一种进一步的方案中,第二反应温度为15℃~35℃。可选地,第二反应温度为23℃~28℃。
[0110]
可选地,第三溶剂选自乙腈、甲基叔丁基醚、乙二醇二甲醚、四氢呋喃或者苯甲醚。
[0111]
可选地,化学式ii所示的化合物的质量:第三溶剂的体积=1g:(6~12)ml。可选地,化学式2所示的化合物的质量:第三溶剂的体积=1g:(8~10)ml。
[0112]
在本发明的一种实施方式中,步骤四
[0113][0114]
在步骤四中,可选地,将化学式p4所示的化合物在第一酸性溶剂中反应。第一酸性溶剂可以选自醋酸、稀盐酸(1~3mol/l)、三氟乙酸或者其他酸性溶剂。
[0115]
可选地,稀盐酸是盐酸水溶液,其浓度为2~3mol/l。
[0116]
可选地,化学式p4所示的化合物的质量:第一酸性溶剂的体积=1g:(2~5)ml。进一步可选地,化学式p4所示的化合物的质量:第一酸性溶剂的体积=1g:(2~3)ml。
[0117]
可选地,化学式p4所示的化合物在第一酸性溶剂中反应时,反应温度为65℃~80℃。进一步可选地,反应温度为70℃~75℃。
[0118]
步骤五:
[0119][0120]
在步骤五中,可选地,将化学式p5所示的化合物、化学式p6所示的化合物和第二酸性溶剂的混合物,在第三反应温度下反应;然后向反应体系中加入三乙酰氧基硼氢化钠并在第四反应温度下反应。
[0121]
进一步可选地,按照摩尔量计,化学式p5所示的化合物:化学式p6所示的化合物=1:(1~1.5)。
[0122]
进一步可选地,第二酸性溶剂包括酸性试剂和第四溶剂。其中,酸性试剂选自醋酸、三氟乙酸、苯磺酸或者其他酸。
[0123]
可选地,按照摩尔量计,化学式p5所示的化合物:酸性试剂=1:(1~2)。进一步可选地,按照摩尔量计,化学式p5所示的化合物:酸性试剂=1:(1~1.2)。
[0124]
可选地,第四溶剂选自醚类溶剂、卤代烷烃类溶剂,例如选自四氢呋喃、甲基四氢呋喃、二氯甲烷、二氯乙烷等。
[0125]
可选地,化学式p5所示的化合物的质量:第四溶剂的体积=1g:(8~12)ml。进一步
可选地,第四溶剂为二氯乙烷,化学式p5所示的化合物的质量:二氯乙烷的体积=1g:10ml。
[0126]
可选地,第三反应温度为40℃~80℃。进一步可选地,第三反应温度为60℃~70℃。
[0127]
可选地,第四反应温度为35℃~50℃。
[0128]
可选地,在加入三乙酰氧基硼氢化钠时,可以将反应体系的温度降低至-10℃~10℃或降温至-5℃~5℃,并分批加入三乙酰氧基硼氢化钠。在加完三乙酰氧基硼氢化钠后,再升温至第四反应温度。
[0129]
本发明涉及了一种双酮类化合物的合成方法,该双酮类化合物是用于制备具有alk5抑制作用的活性咪唑衍生物的关键中间体。按照本发明的合成路线制备活性咪唑化合物,可以避免使用高危险性的试剂氯化氧锆,且减少强酸强碱的使用量,简化后处理过程,减少环境不友好溶剂的使用减少环境污染。因此,本技术的合成方法反应条件和试剂性质比较温和,后处理简便,易于纯化,不需要柱色谱纯化,整个工艺过程符合绿色生产的要求,更适合于工业化生产。
具体实施方式
[0130][0131]
为了更好的明本发明及其所取得的效果,下文中,将参照实施例详细描述本发明双酮类化合物的制备方法和咪唑类衍生物的制备方法做更进一步的说明。然而,根据本说明书的实施例可被修改成各种其他形式,并且本说明书的范围不解释为受限于以下描述的实施例。提供本说明书的实施例是为了更完整地向本领域技术人员描述本说明书。
[0132]
所属领域的专业人员将认识到:本发明所描述的化学反应可以用来合适地制备许多本发明的其他化合物,且用于制备本发明的化合物的其它方法都被认为是在本发明的范围之内。例如,根据本发明那些非例证的化合物的合成可以成功地被所属领域的技术人员通过修饰方法完成,如适当的保护干扰基团,通过利用其他已知的试剂除了本发明所描述的,或将反应条件做一些常规的修改。另外,本发明所申请的反应或已知的反应条件也公认地适用于本发明其他化合物的制备。
[0133]
下面所描述的实施例,除非其他方面表明所有的温度定为摄氏度。试剂购买于商品供应商如aldrich chemical company,arco chemical company and alfa chemicalcompany,使用时都没有经过进一步纯化,除非其他方面表明。一般的试剂从汕头西陇化工厂、广东光华化学试剂厂、广州化学试剂厂、青岛腾龙化学试剂有限公司和青岛海洋化工厂购买得到。原料来自商业采购,供应商例如上海升德医药科技有限公司、上海康拓化工有限公司等。
[0134]
反应一般是在氮气或氩气正压下或在无水溶剂上套一干燥管(除非其他方面表明),反应瓶都塞上合适的橡皮塞。玻璃器皿都是干燥过的。
[0135]
低分辨率质谱(ms)数据的测定条件是:agilent 6120四级杆hplc-m(柱子型号:zorbax sb-c18,2.1
×
30mm,3.5微米,6min,流速为0.6ml/min。流动相:5%-95%(含0.1%甲酸的ch3cn)在(含0.1%甲酸的h2o)中的比例),采用电喷雾电离(esi),在210nm/254nm下,用uv检测。
[0136]
核磁共振氢谱:布鲁克(bruker)400mhz核磁仪,室温条件下,以cdcl3或dmso-d6为
溶剂(以ppm为单位),用tms(0ppm)作为参照标准。当出现多重峰的时候,将使用下面的缩写:s(singlet,单峰)、d(doublet,双峰)、t(triplet,三重峰)、m(multiplet,多重峰)。
[0137]
实施例一:
[0138]
参照如下合成路线2,制备化合物1:
[0139][0140]
步骤1):中间体1-b的合成
[0141]
氮气保护下,向反应瓶中依次加入原料1-a(7.56g,50mmol),s8(19.2g,75mmol),溴乙烷(9.81g,90mmol),nahco3(8.4g,100mmol),tbab(1.62g,5mmol),甲苯100ml,水20ml,搅拌10min,升温至80℃~85℃,保温反应8h,用甲苯(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,产品用1g:4ml的乙醇搅拌0.5h,过滤,抽干,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得1-b中间体(9.52g,收率91%)。
[0142]
在该步骤中,本发明还通过平行实验验证多种不同的反应条件来实现此步骤,例如调整碱的用量、碱的种类、相转移催化剂的种类和用量、溶剂的种类等。
[0143]
通过验证多种不同的反应条件,发现此步骤中的碱(碳酸氢钠)还可以采用碳酸钾、碳酸钠、碳酸氢钾、乙酸钾等常用的无机碱。其中,碱的摩尔量:中间体1-a的摩尔量=1:(1~2)。当采用的碱的碱性越强,则其用量可以越少。在该步骤中,本发明主要采用廉价、易溶的无机碱,以避免有机碱对环境的污染,尤其是减少含有机物的废水排放,提高本发明的方法对环境的友好性。
[0144]
通过验证多种不同的反应条件,发现相转移催化剂(四丁基溴化铵tbab)还可以采用18-冠-6-醚、15-冠-5-醚、tebac(苄基三乙基氯化铵)、tbac(四丁基氯化铵)或者其他相转移催化剂。其中,相转移催化剂的摩尔量:中间体1-b的摩尔量=(0.05~0.2):1。
[0145]
在步骤1)中,当反应规模大时,可以减小相转移催化剂的用量以便节约成本和减少环境污染。示例性地,当中间体1-b的质量大于1kg时,相转移催化剂的摩尔量:中间体1-a的摩尔量=(0.05~0.1):1。
[0146]
在步骤1),反应温度为60℃~85℃,均可顺利进行反应;反应温度为70℃~85℃时反应效率较高,温度高时反应转化速率快。
[0147]
通过平行实验观察多种不同的反应溶剂中的反应收率。为了调整反应原料溶解性,通过对有机溶剂种类和有机溶剂与水的比例进行筛选,原料之间相容性好更有利于反应进行。经过实验发现,本步骤中甲苯和水的混合物作为溶剂时,反应的收率最高。详细实验结果参见表1,表1中公开了多个不同的反应条件及其收率。其中,表1中,各个反应条件之间的差异在于溶剂的种类不同,其他条件(反应原料和底物及用量等)保持一致。
[0148]
表1:不同溶剂实验结果对比
[0149][0150]
从表1中可以看出,当采用甲苯和水的混合物作为溶剂时,收率最高。其次是以苯甲醚和水的混合物作为溶剂时,这可能是由于,当采用亲水溶剂和水的组合时,物料相容性不好而导致反应转化率低。当采用醚和水的组合作为溶剂时,醚类的溶剂极性比甲苯大,导致其反应活性低。
[0151]
步骤2):合成中间体1-d
[0152]
氮气保护下,向反应瓶中依次加入1-b(8.37g,40mmol),原料1-c(10.29g,42mmol),na2co3(8.48g,80mmol),tbab(0.65g,2mmol),甲苯90ml,水30ml,搅拌10min,加入pd132(0.14g,0.2mmol),升温至65℃~70℃,保温反应5h。用甲苯(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.07mpa~-0.08mpa)浓缩至干,用1g:8ml的乙醇加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干得中间体1-d(9.5g,收率89%)。
[0153]
lc-ms(esi,pos.ion)m/z:267.05[m h]
;
[0154]1h-nmr(cdcl3,300mhz)δ(ppm):9.14(s,1h),8.46(s,1h),8.18-8.15(d,1h),8.04-8.01(d,1h),7.88-7.83(m,2h),7.43-7.40(d,1h),2.53(s,3h)。
[0155]
本发明还通过平行反应验证了多种不同的反应条件来实现步骤2),例如调整碱的种类和用量、相转移催化剂的种类和用量、溶剂的种类等。
[0156]
通过验证多种不同的反应条件,发现步骤2)中的碱(碳酸钾)还可以采用碳酸氢钠、碳酸钠、碳酸氢钾、乙酸钾、氢氧化钠等常用的无机碱。其中,中间体1-b:碱的摩尔量=1:(1~2)。在该步骤中,本发明主要采用廉价、易溶的无机碱,以避免有机碱对环境的污染,尤其是减少含有机物的废水排放,提高本发明的方法对环境的友好性。
[0157]
通过验证多种不同的反应条件,发现步骤2)中的相转移催化剂(四丁基溴化铵)还可以采用18-冠-6-醚、15-冠-5-醚、tebac(苄基三乙基氯化铵)、tbac(四丁基氯化铵)或者其他相转移催化剂。其中,相转移催化剂的摩尔量:中间体1-b的摩尔量=(0.05~0.1):1。
[0158]
在步骤2)中,当反应规模大时,可以减小相转移催化剂的用量以便节约成本和减少环境污染。示例性地,当中间体1-c的质量大于1kg时,相转移催化剂的摩尔量:中间体1-c的摩尔量=(0.05~0.07):1。
[0159]
在步骤2),反应温度为60℃~80℃,均可顺利进行反应;反应温度为65℃~75℃时反应效率较高,温度高时反应转化率较高。
[0160]
通过验证多种不同的反应条件,步骤2)中甲苯和水的混合物作为溶剂时,反应的收率最高。参见表2,表2中公开了多个不同的反应条件及其收率。其中,表2中,各个反应条件之间的差异在于溶剂的种类不同,其他条件均一致。
[0161]
表2:不同溶剂实验结果对比
[0162][0163][0164]
从表2数据得出:甲苯和水混合溶剂收率最高,当原料在所选择溶剂中的溶解性不好时,反应转化率低;此外,溶剂的沸点低时反应体系温度较低,反应活性低。
[0165]
步骤3):合成中间体1-e
[0166]
向反应瓶中依次加入中间体1-d(7.99g,30mmol)甲基叔丁基醚80.0ml,搅拌10min,降温至-5℃~0℃,向体系中滴加60%乙二醛二甲缩醛水溶液(9.05ml,60mmol),滴加过程控温-5℃~0℃。滴完后,保温反应1h。分批加入氯化铵(3.54g,66mmol),加完后,室温反应5h,用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩干,得到的固体产品用1g:6ml的乙腈加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得中间体1-e(8.93g,收率85%)。
[0167]
在步骤3)中,氯化铵作为氨供体,还可以采用其他铵盐来替代,例如还可以采用醋酸铵、甲酸铵、碳酸氢铵等铵盐替代。其中,中间体1-d的摩尔量:铵盐的摩尔量=1:(2~2.5)。
[0168]
在步骤3)中,乙二醛二甲缩醛与中间体1-d的摩尔量之比为(2~2.5):1;在本步骤中,乙二醛二甲缩醛溶液滴加过程控温-10℃~10℃,或者-5℃~5℃,从经济性和提高反应转化率角度考虑,优选-5℃~0℃。
[0169]
步骤4):合成中间体1-f
[0170]
氮气保护下,向反应瓶中依次加入中间体1-e(7.01g,20mmol),醋酸20ml,搅拌10min,升温至70℃~75℃,保温反应5h,减压浓缩醋酸,剩余物用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:3ml的石油醚搅拌0.5h,过滤,滤饼真空烘箱(50
℃,-0.08~-0.07mpa)烘干,得中间体1-f(5.78g,收率95%)。
[0171]1h-nmr(dmso-d6,300mhz)δ(ppm):14.18(s,1h),9.79(s,1h),9.56(s,1h),8.55(s,1h),7.98(d,1h,j=9.4hz),7.89-7.73.(m,3h),7.24(s,1h),2.51(s,3h)。
[0172]
在步骤4),反应温度为65℃~80℃,均可顺利进行反应;反应温度为70℃~75℃时反应转化率较高。
[0173]
步骤5:合成化合物1
[0174]
氮气保护下,向反应瓶依次加入中间体1-f(9.12g,30mmol),二氯乙烷45ml,搅拌10min,加入原料1-g(5.0g,45mmol),冰醋酸(2.16g,36mmol),升温至60℃~70℃,保温反应3h,反应体系降温至-5℃~5℃,分批加入nabh(oac)3(12.75,60mmol),加完后升温至40~45℃保温反应20h,用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(150.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:6ml乙醇搅拌0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa)烘干,得化合物1(9.8g,收率82%)。
[0175]
lc-ms(esi,pos.ion)m/z:400.15[m h]
;
[0176]1h-nmr(cdcl3,300mhz)δ(ppm):8.97(s,1h),8.37(s,1h),7.87-7.64(m,2h),7.47(t,1h,j=7.8hz),7.25(d,1h,j=7.9hz),7.15-6.94(m,2h),6.56-6.29(m,3h),4.49(s,2h),2.48(s,3h)。
[0177]
实施例二:
[0178]
参照如下合成路线3,制备化合物2:
[0179][0180]
步骤1:中间体2-a的合成
[0181]
将氢氧化钾和二乙酸碘苯(38.6g,0.12mol)加入到1-(6-叔丁基吡啶-2-基)乙酮(17.7g,0.1mol)的甲醇(150ml)溶液中,然后在室温下搅拌12小时。减压蒸馏(30℃~40℃,-0.09mpa~-0.08mpa)除去溶剂后,在得到的产品中加入水100ml,用二氯甲烷(100ml*2)萃取,有机相用饱和碳酸钠水溶液洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,得到2-a(15.1g,收率78%)。
[0182]
步骤2:中间体2-b的合成
[0183]
氮气保护下,向反应瓶中依次加入原料2-a(9.65g,50mmol),s8(25.6g,0.1mol),溴乙烷(8.18g,75mmol),k2co3(6.9g,50mmol),18-冠-6醚(2.64g,10mmol),甲苯100ml,水20ml,搅拌10min,升温至80℃~85℃,保温反应8h,用甲苯(100.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,产品用1g:3ml的乙醇搅拌0.5h,过滤,抽干,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得2-b中间体(10.06g,收率80%)。
[0184]
步骤3:中间体2-d的合成
[0185]
氮气保护下,向反应瓶中依次加入2-b(7.53g,30mmol),原料1-c(8.09g,33mmol),na2co3(3.18g,30mmol),18-冠-6醚(0.79g,3mmol),甲苯100ml,水30ml,搅拌10min,加入醋酸钯(0.067g,0.3mmol),升温至70℃~75℃,保温反应5h。用甲苯(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,用1g:8ml的乙醇加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干得中间体2-d(7.50g,收率81%)。
[0186]
lc-ms(esi,pos.ion)m/z:309.13[m h]
;
[0187]1h-nmr(cdcl3,300mhz)δ(ppm):9.15(s,1h),8.47(s,1h),8.18-8.15(d,1h),8.03-7.99(d,1h),7.86-7.82(m,2h),7.42-7.39(d,1h),2.54(s,9h)。
[0188]
步骤4:中间体2-e的合成
[0189]
向反应瓶中依次加入中间体2-d(6.16g,20mmol),四氢呋喃50.0ml,搅拌10min,降温至-10℃~-5℃,向体系中滴加60%乙二醛二甲缩醛水溶液(7.55ml,50mmol),滴加过程控温-10℃~0℃。滴完后,保温反应1h。分批加入醋酸铵(3.08g,40mmol),加完后,室温反应5h,用饱和碳酸钠溶液调节ph=7~8,用二氯甲烷(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,得到的固体产品用1g:6ml的乙腈加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得中间体2-e(6.36g,收率81%)。
[0190]
步骤5:中间体2-f的合成
[0191]
氮气保护下,向反应瓶中依次加入中间体2-e(5.5g,14mmol),盐酸22ml,搅拌10min,升温至65℃~70℃,保温反应5h,减压浓缩盐酸,剩余物用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:3ml的石油醚搅拌0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.05mpa)烘干,得中间体2-f(4.23g,收率87%)。
[0192]
步骤6:化合物2的合成
[0193]
氮气保护下,向反应瓶依次加入中间体2-f(4.16g,12mmol),四氢呋喃34ml,搅拌10min,加入原料2-g(1.93g,18mmol),冰醋酸(0.72g,12mmol),升温至40℃~50℃,保温反应3h,反应体系降温至-5℃~5℃,分批加入nabh(oac)3(5.01g,24mmol),加完后升温至35℃~40℃保温反应20h,用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:6ml乙醇搅拌0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa)烘干,得化合物2(3.68g,收率70%)。
[0194]
lc-ms(esi,pos.ion)m/z:438.23[m h]
;
[0195]1h-nmr(cdcl3,300mhz)δ(ppm):8.95(s,1h),8.34(s,1h),7.86-7.67(m,2h),7.48(t,1h,j=7.8hz),7.25(d,1h,j=7.9hz),7.19-6.99(m,2h),6.59-6.30(m,3h),4.51(s,2h),2.53(s,9h),2.35(s,3h)。
[0196]
合成例3:化合物3的合成
[0197][0198]
步骤1:中间体3-a的合成
[0199]
将氢氧化钾和二乙酸碘苯(38.6g,0.12mol)加入到1-(6-乙基吡啶-2-基)乙酮(14.9g,0.1mol)的甲醇(150ml)溶液中,然后在室温下搅拌12小时。减压蒸馏(30℃~40℃,-0.09mpa~-0.08mpa)除去溶剂后,在得到的产品中加入水100ml,用二氯甲烷(100ml*2)萃取,有机相用饱和碳酸钠水溶液洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,得到3-a(11.9g,收率72%)。
[0200]
步骤2:中间体3-b的合成
[0201]
氮气保护下,向反应瓶中依次加入原料3-a(8.25g,50mmol),s8(15.4g,60mmol),溴乙烷(6.5g,60mmol),乙酸钾(7.84g,80mmol),teba(5.70g,2.5mmol),甲苯100ml,水20ml,搅拌10min,升温至80℃~85℃,保温反应8h,用甲苯(100.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,产品用1g:3ml的乙醇搅拌0.5h,过滤,抽干,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得3-b中间体(8.93g,收率80%)。
[0202]
步骤3:中间体3-d的合成
[0203]
氮气保护下,向反应瓶中依次加入3-b(6.69g,30mmol),原料1-c(8.09g,33mmol),nahco3(5.04g,60mmol),teba(0.68g,3mmol),甲苯100ml,水33ml,搅拌10min,加入pd132(,mmol),升温至65℃~70℃,保温反应5h。用甲苯(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(50℃~60℃,-0.09mpa~-0.08mpa)浓缩至干,用1g:8ml的乙醇加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干得中间体3-d(7.15g,收率85%)。
[0204]
lc-ms(esi,pos.ion)m/z:281.10[m h]
;
[0205]1h-nmr(cdcl3,300mhz)δ(ppm):9.16(s,1h),8.44(s,1h),8.17-8.15(d,1h),8.05-8.03(d,1h),7.87-7.82(m,2h),7.44-7.40(d,1h),3.75-3.71(m,2h),2.55-2.52(m,3h)。
[0206]
步骤4:中间体3-e的合成
[0207]
向反应瓶中依次加入中间体3-d(5.6g,20mmol),苯甲醚67.0ml,搅拌10min,降温至5℃~10℃,向体系中滴加60%乙二醛二甲缩醛水溶液(6.63ml,44mmol),滴加过程控温5℃~10℃。滴完后,保温反应1h。分批加入乙酸钾(4.9g,50mmol),加完后,室温反应5h,用饱和碳酸钠溶液调节ph=7~8,用二氯甲烷(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,得到的固体产品用1g:6ml的乙腈加热到50℃~55℃煮洗0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.07mpa)烘干,得中间体3-e(5.61g,收率77%)。
[0208]
步骤5:中间体3-f的合成
[0209]
氮气保护下,向反应瓶中依次加入中间体3-e(5.47g,15mmol),三氟乙酸11ml,搅拌10min,升温至75℃~80℃,保温反应5h,减压浓缩三氟乙酸,剩余物用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:3ml的石油醚搅拌0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa~-0.05mpa)烘干,得中间体3-f(4.11g,收率86%)。
[0210]
步骤6:化合物3的合成
[0211]
氮气保护下,向反应瓶依次加入中间体3-f(3.82g,12mmol),二氯乙烷45ml,搅拌10min,加入原料3-g(2.30g,18mmol),三氟乙酸(2.74g,24mmol),升温至70℃~80℃,保温反应3h,反应体系降温至-5℃~5℃,分批加入nabh(oac)3(5.09g,24mmol),加完后升温至40℃~50℃保温反应20h,用饱和碳酸氢钠溶液调节ph=7~8,用二氯甲烷(100.0ml*2)萃取,有机相用水洗涤,分液,干燥,有机相(40℃~50℃,-0.08mpa~-0.05mpa)浓缩至干,产品用1g:6ml乙醇搅拌0.5h,过滤,滤饼真空烘箱(50℃,-0.08mpa)烘干,得化合物3(3.39g,收率66%)。
[0212]
lc-ms(esi,pos.ion)m/z:430.15[m h]
;
[0213]1h-nmr(cdcl3,300mhz)δ(ppm):8.95(s,1h),8.38(s,1h),7.88-7.64(m,2h),7.43(t,1h,j=7.8hz),7.27(d,1h,j=7.9hz),7.18-6.94(m,2h),6.56-6.30(m,3h),4.50(s,2h),3.78-3.62(m,2h),2.55-2.43(t,3h)。
[0214]
化合物活性测试
[0215]
按照下列测定方法评估本发明合成方法所制备的化合物的alk5激酶磷酸化抑制作用。
[0216]
使用杆状病毒表达系统,使alk5蛋白在sr9昆虫细胞中表达为人重组gst-融合蛋白。利用gsh-琼脂糖(sigma-aldrich)亲和色谱纯化所表达的蛋白质。在来自perkinelmer(boston,ma,usa)的96-孔flashplates
tm
中进行激酶测定,反应体积为50μl。分四个步骤加入反应鸡尾酒试剂,顺序如下:20μl测定缓冲液(标准缓冲液),5μl atp水溶液,5μl含供试化合物的10%的dmso溶液,10μl gsk3(14-27)(200ng)/10μl alk5溶液(1ng)(预混合)。反应鸡尾酒试剂含有60mm hepes-naoh,ph 7.5、3mm mgcl2、3mm mncl2、3μm na3vo4、1.2mm dtt、50μg/ml peg
20000
、1μm[γ-33
p]-atp(大约2.5x105cpm每孔)、200ng/10μl gsk3(14-27)和1ng/10μl alk5。使反应鸡尾酒试剂在30℃下温育60min。用50μl2%(v/v)h3po4终止反应,
将平板抽吸,用200μl 0.9%(w/v)nacl洗涤两次。用beckman coulterbiomek 2000自动系统进行测定。用微量平板闪烁计数器(microbeta,wallac)测定
33
pi的结合(以“cpm”计)。ic
50
定义为在试验条件下,抑制50%酶活性时的化合物浓度。
[0217]
实施例合成的化合物1~化合物4的ic
50
值,如下表所示。
[0218][0219]
根据上表中的测试结果,本技术化合物1~3的alk5激酶抑制ic
50
都小于0.1μm,具有较高的alk5激酶抑制活性。
[0220]
应可理解的是,本技术不将其应用限制到本说明书所列举的实施方式。本技术能够具有其他实施方式,并且能够以多种方式实现并且执行。前述变形形式和修改形式落在本技术的范围内。应可理解的是,本说明书公开和限定的本技术延伸到文中提到或明显的两个或两个以上单独特征的所有可替代组合。所有这些不同的组合构成本技术的多个可替代方面。本说明书所述的实施方式说明了已知用于实现本技术的较优实施方式,并且将使本领域技术人员能够利用本技术。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。