1.本发明涉及有机合成技术领域,特别涉及一种含有取代基的吡嗪环合成方法。
背景技术:
2.早在1879年人们就已经从食品中分离出烷基吡嗪。上个世纪60年代后,随着人 们在大量食品中发现吡嗪类化合物,并且发现这一类化合物是食物中特征香味的重要 因素,吡嗪类化合物逐渐引起了人们的重视。随着研究的深入,人们发现吡嗪类化合 物是重要的有机化工原料及中间体,在医药、农药、香料等领域有着广泛的用途。
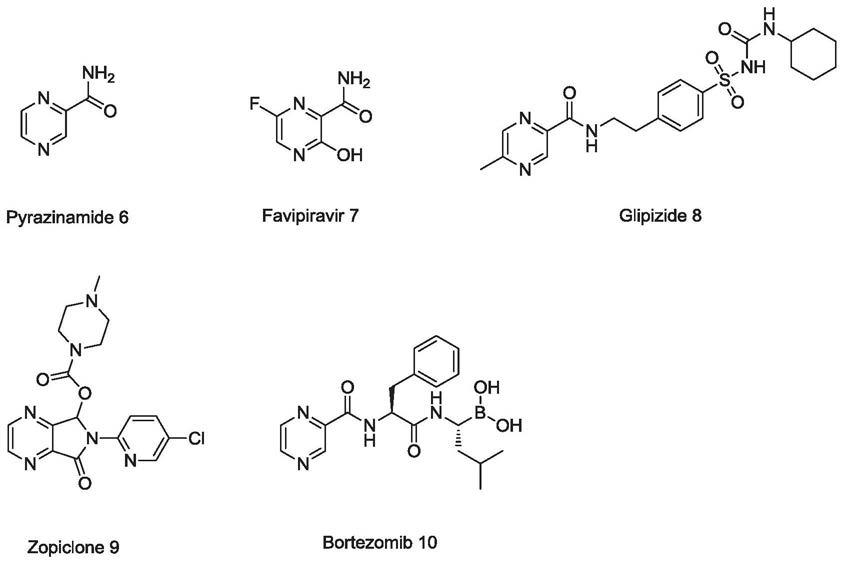
3.具体在医药领域:吡嗪酰胺6对人型结核杆菌有较好的抗菌作用,可用于治疗结 核病;法匹拉韦7作为抗病毒药物,主用于甲型流感的治疗;格列吡嗪8作为口服降 糖药,能促进胰岛素分泌,对大多数二型糖尿病患者有效;佐匹克隆9作为镇静催眠 药,有催眠、镇静、抗焦虑、肌松和抗惊厥等作用;而保特佐米10作为抗肿瘤药物, 可适用于多发性骨髓瘤和套细胞淋巴瘤。
[0004][0005]
鉴于含有取代基的吡嗪环合不仅是重要的活性基团,而且在药物合成中也有着广 泛的应用,研究含有取代基的吡嗪环合的合成方法就具有重要意义。
[0006]
本专利着重研究了含有取代基的吡嗪环合的合成方法,综合查阅文献,发现含有 取代基的吡嗪环合的合成方法主要有下列几条路线:
[0007]
α-(伯氨基)羰基化合物先自我缩合生成相应的二氢吡嗪,再进一步氧化生成相应 的吡嗪。
[0008][0009]
二羰基化合物和二氨基化合物缩合成二氢吡嗪,,再进一步氧化生成相应的吡嗪。
[0010][0011]
喹噁啉或其他稠合吡嗪氧化成吡嗪羧酸。
[0012][0013]
哌嗪脱氢合成吡嗪。有很多报道用不同催化剂通过高温加热使哌嗪反应生成吡嗪。 这一类方法中2,5-二酮哌嗪和三乙基氧鎓四氟硼酸盐反应可生成2,5-二乙氧基-3,6-二氢 吡嗪,经氧化可得到2,5-二乙氧基吡嗪。
[0014][0015]
现有工艺过程存在如下的缺点:(1)原料复杂不易得;(2)反应生成的吡嗪取代 基团有很大的限制;(3)反应得到产物的产率低;(4)反应复杂,操作不便。
技术实现要素:
[0016]
为克服已知路线存在的问题,本发明人设计出新的含有取代基的吡嗪环合成工艺路 线,并通过实验验证其可行性。具体方案如下:
[0017]
一种含有取代基的吡嗪环的合成方法,包括如下步骤:
[0018][0019]
(a)在溶剂中,以式1所示的中草酸类化合物或其盐和式4所示的反式烯二胺类化 合物或其盐为原料缩合得到化合物2;
[0020]
(b)在有机溶剂中,步骤(1)所得化合物2经酰胺化反应得到含有取代基的吡嗪 环化合物3;
[0021]
其中,r1选自氢、c1-c8直链或支链烷基或金属离子;r2选自氢、氰基、氨基、 六元环烷基或苯基,两个r2成环或单独设置。因此化合物4可以为反式烯二胺、二氨基 马来腈或邻苯二胺或者他们的盐。
[0022]
优选的,式1所示化合物选自中草酸、中草酸二乙酯或他们的盐。
[0023]
优选的,式4所示的化合物选自反式烯二胺、二氨基马来腈或邻苯二胺或者他们的 盐。优选的,步骤(a)中,当r1选自氢或金属离子时,所述溶剂为酸性水溶液,当r1选自c1-c8
直链或支链烷基时,所述溶剂为有机溶剂。
[0024]
优选的,当r1选自氢或金属离子时,所述步骤(a)的具体反应步骤为:酸性水 溶液中,0~20℃,式1所示的中草酸类化合物或其盐与式4所示的反式烯二胺类化合物 或其盐或邻苯二胺混合,后控温20~40℃,反应得式2所示化合物;
[0025]
当r1选自c1-c8直链或支链烷基时,所述步骤(a)的具体反应步骤为:有机溶剂 中,0~20℃,式1所示的中草酸类化合物或其盐与式4所示的反式烯二胺类化合物或其 盐或邻苯二胺混合,后升温回流反应,得式2所示化合物。
[0026]
优选的,所述步骤(b)具体步骤为:式2所示化合物在碱性条件下或在催化剂催化 下升温回流反应得到产物。
[0027]
优选的,所述催化剂选自硼酸三(2,2,2-三氟乙基)酯。
[0028]
有益效果
[0029]
本发明提供了一种新的合成路线,原料易得,一般中草酸类化合物均有大量生产; 反式烯二胺类化合物可选择性较多;操作步骤均为常规的化学反应,简易,可操作性强; 最终产品的取代基选择更宽泛;反应步骤短,产品收率高,纯度高。
具体实施方式
[0030]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述, 显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。以下对至 少一个示例性实施例的描述实际上仅仅是说明性的,决不作为对本发明及其应用或使 用的任何限制。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动 前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0031]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根 据本技术的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单 数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含
”ꢀ
和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0032]
在本说明书中,术语“取代”是指在指定原子或基团上的一个或多个氢原子被指定的 基团替代,条件是在现有情况下不超出指定原子的正常化合价。
[0033]
实施例1 3-羟基-2-吡嗪甲酸的制备
[0034]
步骤一:2-((2-胺乙烯基)亚氨基)丙二酸的制备
[0035][0036]
先将11.8g(100mmol)的中草酸加入100ml的1m稀盐酸中,搅拌下分批加入反式烯 二胺盐酸盐10.4g(110mmol),加料过程控温20℃以下。加完控温在20℃保温反应约20 小时。反应完成后,降温,向反应液中滴加20%液碱至ph至6~7,滴加过程控温20℃ 以下。滴加完毕,向反应体系中加入乙酸乙酯200ml,后静置分相,有机相50ml水洗涤。 干燥,过滤,浓缩至干,得到淡黄色油状物14.5g(纯度96%,收率92%)。产物经氢谱分 析,得到其氢谱
数据为:1h nmr(400mhz,dmso)δ8.86(s,2h),7.02(s,2h),6.55(d, 1h),4.28(d,1h).
[0037]
步骤二:3-羟基-2-吡嗪甲酸的制备
[0038][0039]
在250ml反应瓶中加入乙腈100ml,2-((2-胺乙烯基)亚氨基)丙二酸10.0g(63mmol), 硼酸三(2,2,2-三氟乙基)酯38.8g(126mmol),搅拌,升温至回流,保温20小时。待反应完 全,降温至10℃以下,向反应液中加水200ml,滴加盐酸调节ph至3,滴加过程控温 10℃以下。滴完,向反应液中加乙酸乙酯200ml,搅拌分相。有机相经水洗,干燥,过 滤,再加活性炭脱色,过滤,滤液浓缩至约20g,降温结晶得到类白色固体5.3g(纯度99%, 收率60%)。产物经氢谱分析,得到其氢谱数据为:1h nmr(400mhz,dmso)δ9.32(s, 1h),9.02(s,1h),7.12-6.96(m,2h).
[0040]
实施例2 5,6-二氰基-3-羟基-2-吡嗪甲酸的制备
[0041]
步骤一:(z)-2-((2-氨基-1,2-二氰乙烯基)亚氨基)丙二酸二乙酯的制备
[0042][0043]
反应瓶中加入乙醇150ml,17.4g(100mmol)的中草酸二乙酯,二氨基马来腈 11.9g(110mmol),加料过程控温20℃以下。加完升温至回流反应6小时。降温至25℃, 继续保温反应16小时。反应完毕,浓缩至干,得到粗品30.5g,取2.0g进过柱纯化得到 类白色固体1.2(纯度99%,收率69%)。产物经氢谱分析,得到其氢谱数据为:1h nmr(400 mhz,dmso)δ6.78(s,2h),4.16(q,4h),1.25(t,6h).
[0044]
步骤二:5,6-二氰基-3-羟基-2-吡嗪甲酸的制备
[0045][0046]
在50ml反应瓶中加入甲醇20ml,(z)-2-((2-氨基-1,2-二氰乙烯基)亚氨基)丙二酸二乙酯 1.0g(3.8mmol),氢氧化钠0.30g(7.6mmol),搅拌,升温至回流,保温8小时。待反应完 全,降温至10℃以下,向反应液中加水50ml,滴加盐酸调节ph至3,滴加过程控温10℃ 以下。滴完,乙酸乙酯50ml萃取反应液,重复操作三次。合并有机相经水洗,干燥, 过滤,滤液浓缩得到粗品,过柱得到类白色固体0.36g(纯度99%,收率50%)。
[0047]
实施例3 3-羟基喹喔啉-2-羧酸的制备
[0048]
步骤一:2-((2-氨基苯基)亚胺基)丙二酸的制备
[0049][0050]
先将16.2g(100mmol)的中草酸钠盐加入100ml的1m稀盐酸中,搅拌下分批加入邻 苯二胺11.9g(110mmol),加料过程控温20℃一下。加完控温在40℃保温反应约24小时。 反应完成后,降温,向反应液中滴加20%液碱至ph至6~7,滴加过程控温20℃以下。 滴加完毕,向反应体系中加入甲苯200ml,后静置分相,有机相50ml水洗涤。干燥,过 滤,浓缩至干,得到淡黄色固体20.4g(纯度91%,收率98%)。产物经氢谱分析,得到其 氢谱数据为:1h nmr(400mhz,dmso)δ8.36(s,2h),7.20-6.63(m,4h),5.17(s,2h).
[0051]
步骤二:3-羟基喹喔啉-2-羧酸的制备
[0052][0053]
在250ml反应瓶中加入乙腈100ml,2-((2-氨基苯基)亚胺基)丙二酸10.0g(48mmol), 硼酸三(2,2,2-三氟乙基)酯29.6g(96mmol),搅拌,升温至回流,保温20小时。待反应完 全,降温至10℃以下,向反应液中加水200ml,滴加盐酸调节ph至3,滴加过程控温 10℃以下。滴完,向反应液中加甲苯200ml,搅拌分相。有机相经水洗,干燥,过滤, 再加活性炭脱色,过滤,滤液浓缩至约40g,降温结晶得到类白色固体3.1g(纯度93%, 收率32%)。产物经氢谱分析,得到其氢谱数据为:1h nmr(400mhz,dmso)δ9.26(s, 1h),9.00(s,1h),7.36-7.20(m,4h).
[0054]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原 则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。