:
1.本发明属于药物化学领域,具体涉及一种盐酸莫西沙星新的制备方法。
背景技术:
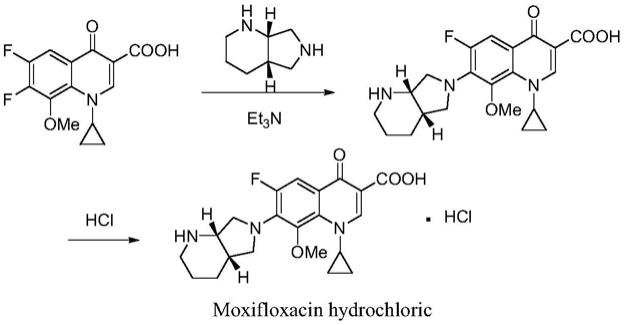
2.盐酸莫西沙星(moxifloxacin hydrochloride)是一种新型的8-甲氧基喹诺酮类抗菌药,化学名为1-环丙基-7-{s,s-2,8-二氮杂双环[4.3.0]壬烷-8-}-6-氟-8-甲氧基-1,4-二氢-4-氧代-3-喹啉羧酸盐酸盐,结构式如下:
[0003][0004]
盐酸莫西沙星是德国bayer公司研发的一款喹诺酮类药物,于1999年9月在德国上市,同年12月在美国上市,2002年在我国批准上市,商品名为拜复乐。临床主要用于社区获得性肺炎(cap)、慢性支气管炎急性发作、泌尿生殖系感染、急性鼻窦炎等。莫西沙星具有抗菌谱广,抗菌活性强,不易产生耐药性,药代动力学性质优良等优点,其临床应用也逐渐增加。
[0005]
盐酸莫西沙星合成,首先母核c7位与支链(s,s)-2,8-二氮杂双环[4.3.0]壬烷发生亲核取代反应,产生莫西沙星,成盐得终产品盐酸莫西沙星。对于连侧链的反应,在反应过程中存在c
7-f与c
6-f的竞争取代,并且母核c8位甲氧基的强推电子效应降低了c
7-f的离去活性,生成c-6位置异构体的副产物,与莫西沙星理化性质相似,难以分离,降低产率。
[0006]
目前对盐酸莫西沙星的合成主要有以下几种工艺路线:
[0007]
(一)欧洲专利ep550903《quinolone and naphthyridione carboxylic acid derivatives as antibacterial agents》公开的盐酸莫西沙星合成方法如下:
[0008][0009]
在三乙胺做碱的条件下,直接将莫西沙星母核和侧链进行缩合。该路线的优点是,直接简单;缺点是,难以控制c6-f与c7-f的竞争取代,形成的c6位副产物,难以分离,并且降低产率。
[0010]
(二)专利ep0464823a1(6,7-substituted-8-alkoxy-1-cyclopropyl-1,4-dihydro-4-oxo-3-quinolinecarboxylic acids-o3,o4)bis(acyloxy-o)borate,and the salts thereof,and methods for their manufacture》和us20060264635《process for preparation of moxifloxacin hydrochloride》公开的盐酸莫西沙星的合成方法如下:
[0011][0012]
硼酸和乙酸酐先形成b(oac)3,b(oac)3和莫西沙星母核形成螯合物,螯合物再与(s,s)-2,8-二氮杂双环[4,3,0]壬烷在三乙胺存在下亲核取代,再用氢氧化钠和乙酸处理得莫西沙星,加盐酸得盐酸莫西沙星。该路线可以避免c-6位竞争取代,但是操作复杂,需要借助螯合物,然后连接侧链连后仍需要解螯合。专利wo2008059223中将乙酸酐换成丙酸酐,形成相应的螯合剂,也可以避免c-6位的位置异构体的副产物,但是仍需要形成螯合物与解螯合。操作复杂。
[0013]
(三)专利ep1832587《method for propreing moxifloxacin and moxifloxacin hydrochloride》公开的盐酸莫西沙星合成方法与上述(二)相似,方法如下:
[0014][0015]
该专利描述中,也是先形成螯合,然后连侧链后再解螯合,只是以三氟化硼为螯合剂。并且文献报道中,是采用一锅法制备盐酸莫西沙星,通过控制温度和加料顺序,实现莫西沙星制备的简化。但喹诺酮羧酸氟螯合物不易形成,反应条件苛刻,需要无水无氧环境并且反应时间长,不适宜工业化生产。
[0016]
(四)专利cn102351858《一种高选择性合成莫西沙星的方法》中,公开了一种盐酸莫西沙星的合成方法如下:
[0017][0018]
该专利中,以硼酸酐和三氟乙酸酐形成的三氟醋酸酐硼酸酯做螯合剂,螯合剂与喹诺酮母核螯合后连接侧链。该路线,原料较贵,反应时间长(26h),并且需要将螯合物分离提纯,然后连接侧链,操作复杂,不利于大规模生产。
[0019]
(五)专利cn103087063《一种莫西沙星及其盐的制备方法》中公开一种盐酸莫西沙星的制备方法如下:
[0020][0021]
该方法与前几种路线相似,也是先用螯合剂形成螯合物,避免c6位的竞争取代,然后与侧链亲核取代。该路线的螯合剂采用三苯基氯化锡,该路线的优点是操作简单,无需将中间体螯合物进行分离,直接加侧链进行亲核取代反应。该路线的缺点是,原料价格较贵,反应时间长(约需16h)。
[0022]
以上合成莫西沙星路线,各有其优缺点,直接用三乙胺连接喹诺酮母核和侧链,则有c6位副产物;用螯合剂可以避免c6位副产物的产生,但是必须先形成螯合物再解螯合,然后解螯合,实验操作复杂,并且收率一般在60-80%之间,即使有文献报道一锅法合成莫西沙星(cn101941969),不需分离螯合物,但也需要硼酸与乙酸酐形成硼酸酯,然后硼酸酯再与喹诺酮母核形成螯合物,反应时间长(8h),浪费乙酸酐,并且需要反复调节温度(80℃-110℃-80℃-rt-83℃),操作还是较复杂,不适合工业化生产。
技术实现要素:
[0023]
本发明的目的是克服现存的莫西沙星合成工艺中操作复杂的弊端,避免分离螯合物和解螯合,简化实验操作,提供一种简洁,反应条件温和,后处理简单,成本低,适合工业化生产和合成路线。
[0024]
本发明所述的莫西沙星制备方法包括如下步骤:
[0025]
1)螯合物的制备:在有机溶剂中,1-环丙基-6,7-二氟-8-甲氧基-4-氧代-1,4-二氢-3-喹啉羧酸与螯合剂形成钨的螯合物;
[0026]
2)盐酸莫西沙星的制备:在有机碱的作用下,步骤1)得到的钨的螯合物与侧链s,s-2,8-二氮杂双环[4,3,0]壬烷反应,得到莫西沙星,莫西沙星加入盐酸成盐,重结晶得盐酸莫西沙星。
[0027]
进一步地,莫西沙星的制备方法如下:
[0028]
1)螯合物的制备:在有机溶剂中,在催化剂氯化锌作用下,1-环丙基-6,7-二氟-8-甲氧基-4-氧代-1,4-二氢-3-喹啉羧酸与螯合剂反应生成钨的螯合物;
[0029]
2)盐酸莫西沙星的制备:在有机碱的作用下,步骤1)得到的钨的螯合物与侧链s,s-2,8-二氮杂双环[4,3,0]壬烷反应,得到莫西沙星,反应液冷却至室温,将溶剂彻底蒸干,加入醇类有机溶剂,过滤钨泥,加入盐酸成盐,重结晶得盐酸莫西沙星。
[0030]
其中,
[0031]
步骤1)中,所述的有机溶剂为乙腈、甲醇、乙醇、异丙醇、丙酮、乙酸乙酯、四氢呋喃、n,n-二甲基亚砜中的一种或几种;优选为乙腈。
[0032]
步骤1)中,温度为50-120℃;更优选为70-90℃。
[0033]
步骤1)中,反应时间为1-6h,更优选2-3h。
[0034]
步骤1)中,所选择的螯合剂为钨酸、二氧化钨、三氧化钨、钨酸钠、钨酸钾、钨酸钙、钨酸钴、钨酸镉、钨酸亚铁、钨酸铵和钨酸锌及碳化钨,更优选为钨酸钠二水合物。
[0035]
步骤1)中,1-环丙基-6,7-二氟-8-甲氧基-4-氧代-1,4-二氢-3-喹啉羧酸与螯合剂的摩尔比为1:0.5-1:3,更优选1:1-1:2。
[0036]
步骤2)中,有机碱为三乙胺、n,n-二异丙基乙胺、环己胺、二乙胺
、
吡啶、4-二甲氨基吡啶、吗啉、n-甲基吗啉中的一种或几种,优选为三乙胺。
[0037]
步骤2)中,温度为50-120℃;更优选为70-90℃。
[0038]
步骤2)中,反应时间为0.5-6h,更优选0.5-1.5h。
[0039]
步骤2)中,1-环丙基-6,7-二氟-8-甲氧基-4-氧代-1,4-二氢-3-喹啉羧酸与有机碱的摩尔比为1:0.5-1:2,更优选1:1.2;1-环丙基-6,7-二氟-8-甲氧基-4-氧代-1,4-二氢-3-喹啉羧酸与侧链s,s-2,8-二氮杂双环[4,3,0]壬烷摩尔比为1:1-1:3;更优选为1:1.5;
[0040]
步骤2)中,所述醇类有机溶剂为甲醇、乙醇、丙醇、异丙醇中的一种或几种,更优选为乙醇;重结晶温度为0-10℃,优选为0℃。
[0041]
以钨酸钠二水合物作为螯合剂为例,本方法的反应路线如下:
[0042][0043]
本发明的创新点:
[0044]
1)与传统的硼鳌合相比,本发明首次以钨化合物做螯合剂,形成螯合物无需进行分离提纯,直接与侧链进行亲核取代,形成莫西沙星也无需再解螯合,经盐酸成盐后,过滤除去钨泥,在醇类溶剂中直接重结晶,得到终产物盐酸莫西沙星,极大的简化了操作步骤,节约了反应试剂,缩短了反应时间,节省了人力。
[0045]
2)反应是一锅法进行直接合成莫西沙星,并且溶剂与温度均无需变化,反应条件温和易操作,绿色环保,其产品纯度高,产率高。
具体实施方式
[0046]
本发明通过以下实施列进一步进行阐述:
[0047]
实施例1:
[0048]
1)螯合物的制备:在1000ml单口圆底烧瓶中加入乙腈300ml,室温下加入1-环丙
基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸30.0g(101.5mmol),钨酸钠二水合物66.8g(202.5mmol),氯化锌0.7g(5.05mmol),逐渐升温至83℃,并保持回流2h,得螯合物。
[0049]
2)盐酸莫西沙星的制备:在上述反应瓶中,保持83℃条件下,加入s,s-2,8-二氮杂双环[4,3,0]壬烷19.2ml(152.2mmol),三乙胺21.1ml(152.2mmol),并保持该回流状态反应2.5h。将反应液冷却至室温,将溶剂减压旋蒸,得黄白色固体。加乙醇600ml,1n hcl调ph=1~2,过滤除去不溶物。0℃冷却析晶。抽滤得黄色固体,乙醇洗涤滤饼2次,将滤饼真空干燥,得黄色固体26g,产率60.5%,纯度99.91%。
[0050]
ms(m/z):[m h]
402.2,[2m h]
803.4.
[0051]1h-nmr(400mhz,dmso-d6)δ:9.89(s,1h),8.77(s,1h),8.67(s,1h),7.70(d,1h,j=14hz),4.12-4.17(m,1h),4.05-4.10(m,1h),3.85-3.90(m,2h),3.73-3.78(m,1h),3.59(s,3h),3.58(s,1h),3.20(s,1h),2.90-2.97(m,1h),2.66(s,1h),1.17-1.83(m,4h),1.16-1.24(m,1h),1.08-1.15(m,1h),0.98-1.06(m,1h),0.83-0.90(m,1h).
[0052]
13
c-nmr(100mhz,dmso-d6)δ:175.95,165.84,153.68,151.20,150.29,140.33,136.72,134.50,117.25,106.37,61.88,54.42,54.02,52.00,41.41,40.63,34.11,20.53,17.53,9.85,8.38.
[0053]
ir(kbr)/cm-1
:3528.0(ν
o-h
),3469.3(ν
n-h
),2927.7(ν-ch
),2796.9(ν-ch
),1709.5(ν
c=o
),1623.2(ν
c=c
),1515.7(ν
c=c
),1455.2(δ
c-h
),1395.4(δ
c-h
),1321.4(ν
c-o
),803.4(δ
ar-h
).
[0054]
实施例2:
[0055]
1)螯合物的制备:在1000ml单口圆底烧瓶中加入乙腈300ml,室温下加入1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸30.0g(101.5mmol),钨酸钠二水合物50.2g(152.2mmol),氯化锌0.7g(5.05mmol),逐渐升温至83℃,并保持回流2h,得螯合物。
[0056]
2)盐酸莫西沙星的制备:在上述反应瓶中,保持83℃条件下,加入s,s-2,8-二氮杂双环[4,3,0]壬烷19.2ml(152.2mmol),三乙胺21.1ml(152.2mmol),并保持该回流状态反应3.5h。将反应液冷却至室温,将溶剂减压旋蒸,得黄白色固体。加乙醇600ml,1n hcl调ph=1~2,过滤除去不溶物。0℃冷却析晶。抽滤得黄色固体,乙醇洗涤滤饼2次,将滤饼真空干燥,得黄色固体25g,纯度99.90%,产率58.2%。
[0057]
实施例3:
[0058]
1)螯合物的制备:在1000ml单口圆底烧瓶中加入乙腈300ml,室温下加入1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸30.0g(101.5mmol),钨酸钾66.0g(202.5mmol),氯化锌0.7g(5.05mmol),逐渐升温至83℃,并保持回流2h,得螯合物。
[0059]
2)盐酸莫西沙星的制备:在上述反应瓶中,保持83℃条件下,加入s,s-2,8-二氮杂双环[4,3,0]壬烷19.2ml(152.2mmol),三乙胺21.1ml(152.2mmol),并保持该回流状态反应2.5h。将反应液冷却至室温,将溶剂减压旋蒸,得黄白色固体。加乙醇600ml,1n hcl调ph=1~2,过滤除去不溶物。0℃冷却析晶。抽滤得黄色固体,乙醇洗涤滤饼2次,将滤饼真空干燥,得黄色固体25.2g,纯度99.91%,产率59.7%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。