1.本发明涉及有机化工技术领域,尤其涉及用于降低亚氨基二乙酸含量的甘氨酸制备方法。
背景技术:
2.甘氨酸(缩写为gly),又称氨基乙酸或乙氨酸,是相对分子质量最小、结构最简单的α-氨基酸,广泛用于农药、化工、医药、日化、食品等行业中。
3.目前,工业上甘氨酸主要以化学合成方法获得,根据工艺不同主要可分为氯乙酸法、施特雷克(strecker)法、改进施特雷克法、直接海因法等。氯乙酸法以氯乙酸、氨为原料,在乌洛托品作用下获得甘氨酸和副产盐氯化铵,该法工艺较短、原料易得,是国内目前主要采用的生产工艺,但该法也具有反应时间长、催化剂乌洛托品消耗大、需要处理大量含盐废水、产品纯度不高等问题,随着国内安全环保要求的提高,该法的进一步发展将会受到限制,且该法获得的甘氨酸纯度较低、纯化困难,对该法获得的甘氨酸在医药、日化、食品等领域的应用也造成了极大限制。国外普遍采用的是改进施特雷克法制备甘氨酸,该法以羟基乙腈为原料,经加氨氨化为氨基乙腈后,在强碱作用下水解获得甘氨酸碱盐,进一步经酸化、浓缩、脱色、分离后获得甘氨酸。该法工艺较为简洁,副产盐硫酸钠较氯乙酸法的氯化铵更易分离,具有一定工业优势,但该法羟基乙腈氨化过程容易产生亚氨基二乙腈、氨基三乙腈等副产物,在后续碱解酸化中生成亚氨基二乙酸(ida)等副产物,需要单独处理。
4.美国专利us4299978公布了一种从甘氨酸母液中分离亚氨基二乙酸的方法,通过加入硫酸,在钠盐存在下,使甘氨酸母液ph为1.5以下,从而使亚氨基二乙酸从母液中结晶出来,避免亚氨基二乙酸的积累对甘氨酸产品造成的影响。该法利用了甘氨酸与亚氨基二乙酸在不同ph下的溶解度实现ida的有效去除,但需要额外使用酸碱调节ph,增大了甘氨酸与硫酸盐分离的处理量,且该方法虽然可以缓解甘氨酸母液中ida的累积,但并没有降低反应体系中甘氨酸、亚氨基二乙酸、硫酸盐的整体分离强度。
5.中国专利cn113563215a公开了一种甘氨酸的生产工艺,包括:s1、氨化;s2、碱解蒸氨;s3、活性炭脱色;s4、离子交换酸化分离:脱色后的甘氨酸盐、亚氨基二乙酸二盐与离子交换树脂上的氢离子发生交换,转化为甘氨酸和亚氨基二乙酸但盐排出系统;s5、甘氨酸浓缩结晶,当循环母液中的亚氨基二乙酸单盐累积到总溶质的10%以上时,将母液抽出;无机盐浓缩结晶,当循环母液溶质中亚氨基二乙酸单盐积累到15%以上含量时,将母液抽出;s6、抽出的母液混合后进行连续色谱分离;s7、亚氨基二乙酸单盐浓缩后加酸酸化结晶回收。该方法通过探索调控亚氨基二乙酸在系统中的最大容纳度实现亚氨基二乙酸的富集和回收,亚氨基二乙酸回收效率相对较高,但存在工艺流程过长、过量富集的亚氨基二乙酸更易带入甘氨酸产品,特别若用作饲料或食品级甘氨酸原料,将提高使用风险。
6.中国专利cn104817466b公开了一种甘氨酸与亚氨基二乙酸联产的方法,通过将含有氨基乙腈和亚氨基二乙腈的混合液,经碱解、酸化,至获得含有甘氨酸和亚氨基二乙酸酸式盐的混合液;将所得的含有甘氨酸和亚氨基二乙酸酸式盐的混合液采用连续色谱分离后
分别得到甘氨酸溶液和含有亚氨基二乙酸酸式盐的溶液;由所得的甘氨酸溶液得到甘氨酸;由所得的含有亚氨基二乙酸酸式盐的溶液得到亚氨基二乙酸。该方法采用连续色谱对甘氨酸和亚氨基二乙酸进行分离,分离较为彻底,且回收后甘氨酸和亚氨基二乙酸二者的循环母液不重合,有利于体系的健康和甘氨酸质量的提升,但由于连续色谱处理浓度低使其分离效率低,加上再生树脂以及反复的洗涤浓缩极大提高能源消耗,特别的,亚氨基二乙酸在甘氨酸中占比并不高,逐次分离会进一步导致其成本堆高,不利于工业化。
7.综上所述,现有技术存在的普遍问题是:主要针对氨基乙腈碱解获得甘氨酸盐和亚氨基二乙酸盐后对其分离过程的改进,且由于甘氨酸、亚氨基二乙酸及无机盐等共存的产物溶液各组分溶解度都较大,常规利用溶解度差的物理分离方法难以有效实现各组分的分离,这导致甘氨酸产品质量受影响,副产物也难以直接利用;采用离交树脂、色谱分离等方法虽然可以提高分离性,但分离过程会大大增加不必要的稀释、洗脱、浓缩、再生等涉及水平衡实现的能源消耗,不利于工业化应用和提高市场竞争力。
技术实现要素:
8.有鉴于此,本发明的目的是提供用于降低亚氨基二乙酸含量的甘氨酸制备方法,解决了现有甘氨酸制备方法中亚氨基二乙酸含量高、难分离的问题。
9.本发明通过以下技术手段解决上述技术问题:
10.用于降低亚氨基二乙酸含量的甘氨酸制备方法,所述制备方法是在羟基乙腈与氨源的氨化过程加入碳源稳定剂,反应获得甘氨酸腈稳定液,甘氨酸腈稳定液再经水解获得甘氨酸;所述甘氨酸腈稳定液中含有海因酸酰胺和/或氨甲酰乙腈,且亚氨基二乙腈占比不高于0.3%。
11.作为优选的,所述甘氨酸腈稳定液的制备如下:
12.所述碳源稳定剂与氨源预混合后,再与羟基乙腈混合进行氨化反应,获得甘氨酸腈稳定液;
13.或者所述碳源稳定剂与羟基乙腈预混合后,再与氨源混合进行氨化反应,获得甘氨酸腈稳定液;
14.或者所述羟基乙腈与氨源预混合后,再与碳源稳定剂混合进行氨化反应,获得甘氨酸腈稳定液。
15.作为优选的,所述氨化反应中控制温度为55~130℃,时间为1~4min。
16.作为优选的,所述氨化反应分两阶段进行,分别为一阶段55℃反应2min,二阶段100~130℃反应1~2min。
17.作为优选的,所述碳源稳定剂为二氧化碳、碳酸氢铵、碳酸铵、尿素中的至少一种。
18.作为优选的,所述氨源选自氨水、液氨、氨气中至少一种。
19.作为优选的,所述羟基乙腈与氨源中的氨的摩尔比为1:3~5,所述羟基乙腈与碳源稳定剂的摩尔比为1:0.05-0.2。
20.作为优选的,所述甘氨酸腈稳定液的水解反应过程中需加入强碱,反应获得的碱解液进行脱氨、脱色、无机酸酸化处理,得到的含甘氨酸的水溶液经浓缩、结晶得到甘氨酸晶体;
21.所述强碱为氢氧化钠、氢氧化钾、氢氧化钙中的至少一种,所述无机酸为硫酸、盐
酸、磷酸中的至少一种。
22.作为优选的,所述甘氨酸腈稳定液的水解反应过程中加入强酸,反应获得的酸解液进行脱氨、浓缩、结晶处理,得到甘氨酸;所述强酸为硫酸。
23.作为优选的,所述甘氨酸腈稳定液的水解反应过程中加入固体催化剂,催化水解获得的催化水解液分离固体催化剂后,经脱氨、浓缩、结晶获得甘氨酸晶体;
24.所述固体催化剂为锆、锌、钛、锰、铜、铁、铂、铈、铌、铬、钼的氧化物中的至少一种。
25.本发明的甘氨酸制备方法,通过在羟基乙腈氨化过程加入碳源稳定剂,可以降低甚至避免氨化过程亚氨基二乙腈的生成,获得基本不含亚氨基二乙腈的甘氨酸腈稳定液主要成分为氨基乙腈、海因酸酰胺和/或氨甲酰乙腈,均可在后续反应中被转化为甘氨酸,从而避免了亚氨基二乙酸的生成以及伴随而来的分离难题。特别在选用尿素作为碳源稳定剂时,可通过优先将尿素与羟基乙腈混合后,再与氨源进行氨化反应,可大大减少尿素添加量,从而可通过少量的碳源稳定剂添加实现甘氨酸腈的稳定和亚氨基二乙腈的抑制,经济高效。
附图说明
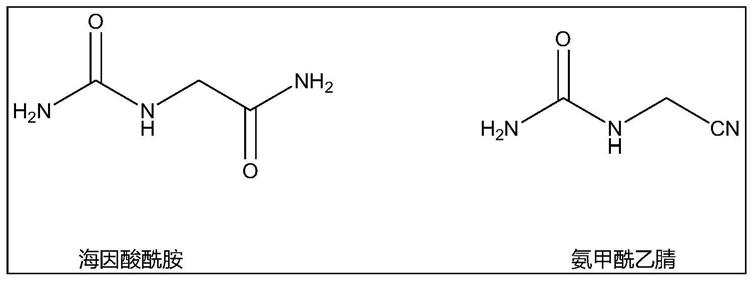
26.图1是海因酸酰胺、氨甲酰乙腈的结构式。
具体实施方式
27.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
28.本发明的用于降低亚氨基二乙酸含量的甘氨酸制备方法,在羟基乙腈与氨源的氨化过程加入碳源稳定剂,反应获得甘氨酸腈稳定液,甘氨酸腈稳定液再经水解获得甘氨酸;所述甘氨酸腈稳定液中含有海因酸酰胺和/或氨甲酰乙腈,且亚氨基二乙腈占比不高于0.3%。其中,甘氨酸腈稳定液的制备既可以是所述碳源稳定剂与氨源预混合后,再与羟基乙腈混合进行氨化反应,获得甘氨酸腈稳定液;又可以是碳源稳定剂与羟基乙腈预混合后,再与氨源混合进行氨化反应,获得甘氨酸腈稳定液,尤其是选用尿素作为碳源稳定剂时,先将尿素与羟基乙腈混合再与氨源混合进行氨化反应,发现尿素提前与羟基乙腈混合其稳定效果(亚氨基二乙腈含量少)更好;还可以是羟基乙腈与氨源预混合后,再与碳源稳定剂混合进行氨化反应,获得甘氨酸腈稳定液;再或者是碳源稳定剂与羟基乙腈、氨源混合进行氨化反应,获得甘氨酸腈稳定液。其中,氨化反应中控制温度为55~130℃,时间为1~4min,进一步,氨化反应分两阶段进行,分别为一阶段55℃反应2min,二阶段100~130℃反应1~2min。
29.其中的碳源稳定剂为二氧化碳、碳酸氢铵、碳酸铵、尿素中的至少一种,氨源选自氨水、液氨、氨气中至少一种,优选10-30w%氨水,羟基乙腈优选使用氢型离子交换树脂处理至ph=2-5的羟基乙腈。羟基乙腈与氨源中的氨的摩尔比为1:3~5,所述羟基乙腈与碳源稳定剂的摩尔比为1:0.05-0.2。
30.甘氨酸腈稳定液的水解反应过程中可以加入强碱,反应获得的碱解液进行脱氨、
脱色、无机酸酸化处理,得到的含甘氨酸的水溶液经浓缩、结晶得到甘氨酸晶体;其中的强碱为氢氧化钠、氢氧化钾、氢氧化钙中的至少一种,所述无机酸为硫酸、盐酸、磷酸中的至少一种。
31.甘氨酸腈稳定液的水解反应过程中还可以加入强酸,反应获得的酸解液进行脱氨、浓缩、结晶处理,得到甘氨酸;其中的强酸为硫酸。
32.甘氨酸腈稳定液的水解反应过程中还可以加入固体催化剂,催化水解获得的催化水解液分离固体催化剂后,经脱氨、浓缩、结晶获得甘氨酸晶体;其中的固体催化剂为锆、锌、钛、锰、铜、铁、铂、铈、铌、铬、钼的氧化物中的至少一种。
33.为了更好的了解本发明的甘氨酸制备方法,进行了以下实施例的实验。
34.第一部分甘氨酸腈稳定液制备
35.实施例1(加入0.2当量碳酸氢铵;以羟基乙腈计,下同)
36.将31.93g碳酸氢铵(99w%)碳源稳定剂与680g氨水(25w%)混合得氨源液流,量取175.5g羟基乙腈(65w%,ph=4)作为羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在115℃,压力1.3mpa条件下进行氨化反应,反应停留时间为4分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示未检测出明显的亚氨基二乙腈,产物中氨基乙腈占比82.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比17.2%。海因酸酰胺、氨甲酰乙腈的结构式如图1所示。
37.实施例2(加入0.2当量二氧化碳)
38.通入17.6g二氧化碳气体(0.4mol)碳源稳定剂与680g氨水(25w%)混合得氨源液流,量取175.5g羟基乙腈(65w%,ph=4)作为羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在115℃,压力1.3mpa条件下进行氨化反应,反应停留时间为3分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示亚氨基二乙腈占比约0.2%,产物中氨基乙腈占比86.4%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比13.3%。
39.实施例3(加入0.15当量尿素)
40.将18.2g尿素(99w%)碳源稳定剂与680g氨水(25w%)混合得氨源液流,量取175.5g羟基乙腈(65w%,ph=2)作为羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在130℃,压力1.8mpa条件下进行氨化反应,反应停留时间为1分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示未检测出明显的亚氨基二乙腈,产物中氨基乙腈占比92.2%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比7.6%。
41.对比实施例1(不加碳源稳定剂)
42.将680g氨水(25w%)混合得氨源液流,量取175.5g羟基乙腈(65w%,ph=4)作为羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在115℃,压力1.3mpa条件下进行氨化反应,反应停留时间为4分钟,反应结束管式反应器出口获得甘氨酸腈反应液,hplc分析显示亚氨基二乙腈占比5.83%,氨基乙腈占比94.17%。
43.实施例4(二段反应加入0.1当量碳酸氢铵)
44.取680g氨水(25w%)和175.5g羟基乙腈(65w%,ph=2)分别作为氨源液流和羟基
乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入一段管式反应器中,在55℃停留反应2分钟,与52.7g碳源稳定剂(30w%碳酸氢铵水溶液)混合进入二段管式反应器,在100℃进行二段氨化反应,反应停留时间为2分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示未检测出明显的亚氨基二乙腈,产物中氨基乙腈占比82.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比17.2%。
45.实施例5(二段反应加入0.1当量碳酸铵)
46.取680g氨水(25w%)和175.5g羟基乙腈(65w%,ph=2)作为氨源液流和羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入一段管式反应器中,在55℃停留反应2分钟,与64g碳源稳定剂(30w%碳酸铵水溶液)混合进入二段管式反应器,在130℃进行二段氨化反应,反应停留时间为1分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示未检测出明显的亚氨基二乙腈,产物中氨基乙腈占比83.3%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比16.6%。
47.实施例6(0.05当量尿素与羟基乙腈混合)
48.将6.1g尿素(0.1mol)碳源稳定剂与175.5g羟基乙腈(65w%,ph=4)混合作为羟基乙腈液流,称取680g氨水(25w%)作为氨源液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在120℃,压力1.3mpa条件下进行氨化反应,反应停留时间为2分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示未检测出明显的亚氨基二乙腈,产物中氨基乙腈占比90.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比9.4%。
49.实施例7(0.1当量二氧化碳与氨气混合)
50.将17.6g二氧化碳(约0.4mol)碳源稳定剂与102g氨气(6mol)混合得氨源液流,量取175.5g羟基乙腈(65w%,ph=4)作为羟基乙腈液流,分别通过物料泵将氨源液流与羟基乙腈液流泵入静态混合器内混合后送入管式反应器中,在130℃,压力1.5mpa条件下进行氨化反应,反应停留时间为1分钟,反应结束管式反应器出口获得甘氨酸腈稳定液,hplc分析显示亚氨基二乙腈占比约0.27%,产物中氨基乙腈占比92.86%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比6.92%。
51.通过实施例1-7及对比实施例1实验结果可知,在碳源稳定剂存在下羟基乙腈氨化获得的甘氨酸腈稳定液,相较羟基乙腈直接氨化获得的甘氨酸腈反应液,氨化副产物亚氨基二乙腈大大降低(不高于0.3%),而现有技术羟基乙腈直接氨化反应获得的甘氨酸腈反应液中亚氨基二乙腈含量在5.83%,亚氨基二乙腈在后续反应会转化为不利的亚氨基二乙酸,因此本发明在大大抑制亚氨基二乙腈产生的情况下,可以有效解决后续甘氨酸生产中亚氨基二乙酸的不利影响。
52.第二部分:甘氨酸腈稳定液制备甘氨酸
53.实施例8(甘氨酸腈碱解,酸化)
54.将实施例1获得的甘氨酸腈稳定液(氨基乙腈占比82.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比17.2%)转入2l锆材高压釜(316l),加入91.7g氢氧化钠(96%),在85℃碱解40分钟,获得碱解液867.7g,hplc分析甘氨酸含量17.15%,未检测出亚氨基二乙酸,以羟基乙腈计收率约99.1%。
55.所得碱解液经汽提脱氨后游离氨低于100ppm,脱氨后碱解液加入7.5g活性炭
(0.5%产品质量比)在60℃搅拌脱色45min获得脱色碱解液,在搅拌下继续向脱色碱解液中加入343g硫酸溶液(30w%,)酸化,获得甘氨酸与硫酸钠混合液,浓缩结晶获得甘氨酸固体产品纯度98.2%,重结晶一次后甘氨酸固体产品纯度达99.9%。
56.实施例9(甘氨酸腈直接酸解)
57.将实施例3获得的甘氨酸腈稳定液(氨基乙腈占比92.2%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比7.6%)经闪蒸脱除部分游离氨后转入2l锆材高压釜(316l),向开启搅拌的高压釜中加入350g硫酸溶液(70w%),密闭高压釜在85℃酸解90min,获得酸解液1064.7g,hplc分析甘氨酸含量14.02%,未检测出亚氨基二乙酸,以羟基乙腈计收率约99.4%。
58.实施例10(甘氨酸腈氧化铈催化)
59.将实施例6获得的甘氨酸腈稳定液(氨基乙腈占比90.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比9.4%)转入2l锆材高压釜(316l),加入344g二氧化铈(平均粒径约50nm,八面体晶型),在50℃催化水解60分钟,分离固体催化剂后获得液相为催化水解液797.6g,hplc分析甘氨酸含量18.77%,未检测出亚氨基二乙酸,甘氨酸以羟基乙腈计收率约99.7%。
60.将上述催化水解液直接脱氨、浓缩、结晶获得甘氨酸晶体,分析纯度98.6%,在纯水中重结晶后纯度达99.9%。
61.实施例11(甘氨酸腈氧化铈 氧化锆催化)
62.将实施例4获得的甘氨酸腈稳定液(氨基乙腈占比82.6%,海因酸酰胺、氨甲酰乙腈及其他稳定成分占比17.2%)转入2l锆材高压釜(316l),加入290.1g二氧化锆 二氧化铈混合催化剂(氧化锆与氧化铈组分比为0.5:1,与羟基乙腈摩尔比1:1,平均粒径约50nm,二氧化铈晶型为八面体),在50℃催化水解60分钟,分离固体催化剂后获得液相为催化水解液903.8g,hplc分析甘氨酸含量16.53%,未检测出亚氨基二乙酸,甘氨酸以羟基乙腈计收率约99.7%。将催化水解液直接脱氨、浓缩、结晶获得甘氨酸晶体,分析纯度98.8%,在纯水中重结晶后纯度达99.9%。
63.对比实施例2(甘氨酸腈碱解,酸化)
64.将对比施例1获得的甘氨酸腈反应液(亚氨基二乙腈占比2.13%,氨基乙腈占比97.65%)转入2l锆材高压釜(316l),加入91.7g氢氧化钠(96%),在85℃碱解40分钟,获得碱解液859.4g,hplc分析甘氨酸含量15.13%,亚氨基二乙酸1.64%,甘氨酸以羟基乙腈计收率约86.64%,甘氨酸与亚氨基二乙酸以羟基乙腈计收率约97.14%。
65.所得碱解液经汽提脱氨后游离氨低于100ppm,脱氨后碱解液加入7.5g活性炭(0.5%产品质量比)在60℃搅拌脱色45min获得脱色碱解液,在搅拌下继续向脱色碱解液中加入343g硫酸溶液(30w%,)酸化,获得甘氨酸与硫酸钠混合液,浓缩结晶获得甘氨酸固体产品纯度95.8%,重结晶一次后甘氨酸固体产品纯度达98.9%。
66.由实施例8-11与对比实施例2可知,本发明获得的包含海因酸酰胺和/或氨甲酰乙腈的甘氨酸腈稳定液可以稳定高效地转化为目标产物甘氨酸(转化率99%以上),而现有技术无论是甘氨酸转化率还是甘氨酸加亚氨基二乙酸的总收率均低于本发明实施方案,另外本发明获得的甘氨酸产品纯度较现有技术(见对比实施例2)也更高。
67.以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发
明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。本发明未详细描述的技术、形状、构造部分均为公知技术。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。