1.本发明涉及机合成技术领域,尤其是一种手性四氢咔唑类多环衍生物及其在抗肿瘤相关药物的应用。
背景技术:
2.手性四氢咔唑在许多天然产物(如生物碱)中发现的具有重要生物活性的多环吲哚结构,这些化合物是有用的活性分子结构单元,应用于生物药物和功能材料,此外,含咔唑单元的天然产物具有广泛的生物学特性。是近些年来咔唑衍生物化学研究的一个热点。其中,含咔唑框架的合成更是引起了化学家们的广泛关注。令人惊讶的是,在过去50年中,数十种合成方法已经开发出来,例如:近年来,macmillan,melchiorre和chen、zanardi和其他的研究小组报道了通过非过渡金属方法对手性咔唑及其衍生物的对映选择性催化反应,该反应受到了广泛的关注。在这些方法中最成功的方法是使用手性胺催化剂通过亚胺或烯胺的催化途径活化反应原料。合成此类分子的一种有前景的方法可以通过吲哚衍生物的功能化来实现。在2013中,池组报道了可以通过n-杂环卡宾(nhc)有机催化活化吲哚-3-甲醛,形成邻醌二甲烷中间体,与各类取代的三氟苯乙酮形成[4 2] 反应。鉴于我们课题组前期的工作,我们设计了类似的吲哚邻二喹甲烷中间体与 nhc结合的亲二烯物反应可以有效地合成带有与吲哚骨架融合的全碳环的咔唑。
[0003]
尽管许多课题组报道了一类咔唑类的合成方法,但是由于其在合成中需要使用昂贵的金属催化剂、以及反应需要无水无氧氮气保护下进行,条件比较苛刻,实际应用性差。
技术实现要素:
[0004]
本发明的目的是:提供一种手性四氢咔唑类多环衍生物及其制备方法与应用,它方法具有操作简便,试剂易得和条件温和等优点,合成了复杂的手性四氢咔唑。
[0005]
本发明还发现该类化合物在制备防治肿瘤疾病药物中的应用。
[0006]
本发明是这样实现的:手性四氢咔唑类多环衍生物,该化合物具有如下结构通式:
[0007][0008]
其中r1为5-ch3、5-och3,或5-cl;r2为2-ch3、2-och3、4-ch3、4-och3或4-br;x为o、n或s;r3为boc或ts。
[0009]
手性四氢咔唑类多环衍生物的制备方法,包括如下步骤:
[0010]
(1)2-甲基吲哚-3-甲醛的合成:
[0011]
在氮气保护条件下,将反应装置置于冰浴条件下,向反应器中加入dmf,然后逐滴
加入三氯氧磷,冰浴条件下继续搅拌30分钟后,将不同取代基的2-甲基吲哚溶于dmf中滴加至体系,继续搅拌一小时,反应结束,将反应体系倒入冰水中,有乳白色的固体析出,抽滤、水洗涤,干燥,获得中间体i;
[0012]
(2)3-醛基-2-甲基-n-甲酸叔丁酯-吲哚的合成:
[0013]
将步骤(1)制得的中间体i溶于乙腈溶液中,加入二碳酸二叔丁酯,然后 dmap,室温搅拌30min,tlc跟踪反应进程,待反应结束,柱色谱纯化(洗脱剂:pe:ea=10:1)得到中间体ii。;
[0014]
(3)3-(2,2-二氰基乙烯基)-2-甲基-1h-吲哚-1-甲酸叔丁酯的合成:
[0015]
将步骤(2)制得的中间体ii溶于无水乙醇中,加入丙二腈,搅拌均匀后,加入et3n,继续常温搅拌,tlc点板跟踪至中间体ii完全转化,有固体析出,抽滤,滤饼少量无水乙醇洗涤,烘干,制得各种取代的目标原料;
[0016]
上述反应路线如下:
[0017][0018]
其中r1为5-ch3、5-och3,或5-cl。
[0019]
具体合成例举如下:
[0020][0021]
向配备有磁力搅拌棒的干燥小瓶中添加亚甲基丙二腈1a(0.10mmol),α
‑ꢀ
溴代烯醛2a(0.30mmol),前催化剂盐a(0.02mmol)和et3n(0.3mmol)。加入干燥的thf(2ml),然后将反应混合物在常温下搅拌直至1a完全消耗(通过tlc监测)。将混合物在减压下浓缩,通过柱色谱法(石油醚/etoac=5: 1)纯化得到所需产物3a,其通过1h nmr,
13
c nmr谱确认其结构,对映比率通过手性hplc测定。
[0022]
所述的步骤(1)中dmf、三氯氧磷和2-甲基吲哚的物质的量之比为3:3:1。
[0023]
所述步骤(2)中二碳酸二叔丁酯、dmap与中间体i的量之比为1.2:0.2:1.0。
[0024]
所述步骤(3)中丙二腈、三乙胺与中间体ii的物质的量之比为1.2:0.2:1.0。
[0025]
手性四氢咔唑类多环衍生物在药物合成中的应用。
[0026]
本发明的有益效果:本发明所提供的手性四氢咔唑合成方法具有操作简便、原料易得、条件温和和易于制备、底物普适性广等优点。所获得的手性四氢咔唑可以与烯烃类化合物发生sonogashira偶联反应,可以发生氧化、还原、水解、胺解等反应,转化过程中活性高,产率高、立体选择性高等优点。
附图说明
[0027]
图1为本发明合成原理示意图。
具体实施方式
[0028]
实施例1
[0029]
本实施例1的手性四氢咔唑3a的制备方法如下:
[0030]
(1)3a的合成:吲哚-2-甲基-亚甲基丙二腈1a(0.05mmol)、α-溴代烯醛2a(0.15mmol)、前手性催化剂盐a(0.01mmol)、ms(50mg)和et3n(0.15mmol)。加入新鲜蒸馏的thf(1ml),然后将反应混合物在常温下搅拌24小时直至1a完全消耗(通过tlc监测)。待反应完全后,浓缩有机相,石油醚/乙酸乙酯为洗脱剂色谱柱分离得到所需产物3a,其通过1hnmr,
13
cnmr谱确认结构,其对映比率通过手性hplc测定,产率:73%;1hnmr(400mhz,cdcl3):δ=8.16-8.14(m,1h),7.84-7.82(m,1h),7.67-7.65(m,3h),7.45-7.34(m,10h),5.64(s,1h),4.34(d,j=4.2hz,1h),3.79(dd,j1=18.6hz,j2=5.3hz,1h),3.73-3.66(m,1h),3.54(dd,j1=18.6hz,j2=5.3hz,1h),3.41-3.33(m,1h),1.68(s,9h)ppm;
13
cnmr(100mhz,cdcl3):δ=166.65,150.10,138.33,137.65,136.08,135.04,133.73,129.39,129.32,128.54,128.47,127.57,126.96,125.12,123.84,118.81,116.04,113.67,113.33,112.68,110.90,85.22,79.82,46.18,41.75,41.04,40.68,34.54,28.25;hrms(esi,m/z):calcd.forc
36h31
brn3o
4
:m/z=648.1492,found:m/z=648.1485;hplcanalysis:99%ee(chiralpakiccolumn,254nm,90:10hexanes/iproh,1.0ml/min),rt(minor)=12.9min,rt(major)=21.7min.
[0031]
实施例2
[0032]
本实施例2的手性四氢咔唑3b的制备方法如下:
[0033]
3b的合成:同实施例1中3a的合成;5-甲基吲哚-2-甲基-亚甲基丙二腈1b(0.05mmol)、α-溴代烯醛2a(0.15mmol)、前手性催化剂盐a(0.01mmol)、ms(50mg)和et3n(0.15mmol)。加入新鲜蒸馏的thf(1ml),然后将反应混合物在常温下搅拌24小时直至1b完全消耗(通过tlc监测)。待反应完全后,浓缩有机相,石油醚/乙酸乙酯为洗脱剂色谱柱分离得到所需产物3b,其通过1hnmr,
13
cnmr谱确认结构,其对映比率通过手性hplc测定,产率:73%,白色固体,1hnmr(400mhz,cdcl3):δ=8.01(d,j=8.6hz,1h),7.66-7.61(m,4h),7.44-7.32(m,8h),7.20(d,j=8.6hz,1h),5.63(s,1h),4.32(d,j=4.4hz,1h),3.80(dd,j1=18.6hz,j2=5.3hz,1h),3.71-3.64(m,1h),3.55(dd,j1=12.2hz,j2=4.6hz,1h),3.38-3.31(m,1h),2.48(s,3h),1.66(s,9h);
13
cnmr(100mhz,cdcl3):δ=166.7,150.1,138.4,137.6,135.1134.3,133.8,133.5,129.4,129.3,128.5,127.6,127.2,126.5,118.8,115.7,113.7,113.5,112.7,110.7,85.0,79.8,46.2,41.8,41.1,40.7,34.5,28.3,21.5;hrms(esi,m/z):calcd.forc
37h33
brn3o
4
:m/z=662.1649,found:m/z=662.1661;hplcanalysis:98%ee(chiralpakiccolumn,254nm,90:10hexanes/iproh,1.0ml/min),rt(minor)=11.2min,rt(major)=15.4min.
[0034]
实施例3
[0035]
本实施例3的手性四氢咔唑3c的制备方法如下:
[0036]
3c的合成:同实施例1中3a的合成;5-甲氧基吲哚-2-甲基-亚甲基丙二腈1c(0.05mmol)、α-溴代烯醛2a(0.15mmol)、前手性催化剂盐a(0.01mmol)、ms(50mg)和et3n(0.15mmol)。加入新鲜蒸馏的thf(1ml),然后将反应混合物在常温下搅拌24小时直至1c完
全消耗(通过tlc监测)。待反应完全后,浓缩有机相,石油醚/乙酸乙酯为洗脱剂色谱柱分离得到所需产物3c, 其通过1h nmr,
13
c nmr谱确认结构,其对映比率通过手性hplc测定,产率: 79%,白色固体。1h nmr(400mhz,cdcl3):δ=8.02 (d,j=9.3hz 1h),7.65-7.61(m,3h),7.43-7.32(m,9h),6.98-6.95(m,1h),5.62 (s,1h),4.33(d,j=4.7hz,1h),3.87(s,3h),3.80(dd,j1=18.6hz,j2=5.1hz,1h), 3.71-3.64(m,1h),3.57(dd,j1=12.2hz,j2=4.5hz,1h),3.39-3.32(m,1h),1.66 (s,9h);
13
c nmr(100mhz,cdcl3):δ=166.7,156.6,150.0,138.3,138.1,134.9, 133.7,130.5,129.3,129.3,128.5,128.5,127.7,127.6,117.0,114.3,114.0,113.4, 112.8,110.8,100.9,85.0,79.8,55.7,46.2,41.8,41.2,40.6,34.6,28.3;hrms(esi, m/z):calcd.for c
37h33
brn3o
5
:m/z=678.1598,found:m/z=678.1622;hplc analysis:97%ee(chiralpak ic column,254nm,90:10hexanes/iproh,1.0 ml/min),rt(minor)=15.1min,rt(major)=32.9min.
[0037]
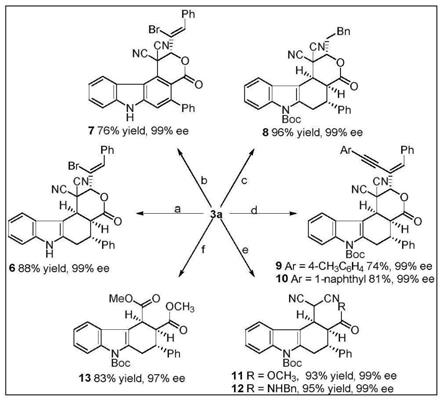
利用该方法制备的手性咔唑及其衍生物如表1所示:
[0038]
表1已合成的手性四氢咔唑及其衍生物的结构
[0039]
[0040]
[0041]
7.16-7.07(m,2h),6.29(s,1h),4.85(d,j=5.2hz,1h),4.0-3.9(m,1h),3.70
‑ꢀ
3.64(m,1h),3.29-3.14(m,2h);13c nmr(100mhz,dmso):δ=167.7,140.9, 136.8,136.7,135.9,134.3,129.8,129.6,129.1,128.9,128.7,127.9,126.5,121.9, 119.9,118.7,116.6,115.4,111.9,104.1,77.5,46.3,43.0,38.8,31.0;hrms(esi, m/z):calcd.for c31h23brn3o2 :m/z=548.0968,found:m/z=548.0989;hplc analysis:99%ee(chiralpak ic column,254nm,95:5hexanes/iproh,1.0 ml/min),rt(major)=33.3min,rt(minor)=39.3min.
[0046]
实施例5
[0047]
邻硝基碘苯与丙烯醛偶联制备化合物7e:10ml反应瓶中,称入1mol%醋酸钯与2mol%二茂铁基嘧啶类多齿配体6h,加入6ml水,搅拌5min后,一次加入2mmol邻硝基碘苯、2.4mmol丙烯醛、3mmol磷酸钾和0.4mmol四丁基溴化铵,升温至40℃反应至反应完全(约2h),加入30ml乙酸乙酯,水洗三次,合并有机相,无水硫酸钠干燥,浓缩,剩余物通过柱色谱分离,pe/ea=10:1 为洗脱剂,得白色棉花状固体332mg,收率94%。1h nmr(300mhz,cdcl3)δ 9.76(d,j=7.5hz,1h),8.03-8.11(m,2h),7.59-7.75(m,3h),6.59-6.67 (m,1h)ppm;
13
c nmr(75mhz,cdcl3)δ194.0,148.4,147.6,135.0, 133.0,131.5,130.4,129.5,125.6ppm。
[0048]
实施例6
[0049]
对氯碘苯与丙烯酸丁酯偶联制备化合物7n:10ml反应瓶中,称入1mol%醋酸钯与2mol%二茂铁基嘧啶类多齿配体6h,加入6ml水,搅拌5min后,一次加入2mmol对氯碘苯、2.4mmol丙烯酸丁酯、3mmol磷酸钾和0.4mmol四丁基溴化铵,升温至40℃反应至反应完全(约3h),加入30ml乙酸乙酯,水洗三次,合并有机相,无水硫酸钠干燥,浓缩,剩余物通过柱色谱分离, pe/ea=15:1为洗脱剂,得淡黄色固体442mg,收率93%。1hnmr(300mhz, cdcl3)δ7.61(d,j=15.6hz,1h),7.44(d,j=8.7hz,2h),7.35(d,j =8.7hz,2h),6.40(d,j=15.9hz,1h),4.21(t,j=6.6hz,2h),1.50-1.69 (m,2h),1.38-1.44(m,2h),0.96(t,j=7.2hz,3h)ppm;
13
cnmr(75mhz, cdcl3)δ167.0,143.1,136.1,133.0,129.2,129.1,118.9,64.5,30.7, 19.2,13.7ppm。.
[0050]
实施例7
[0051]
对硝基氯苯与丙烯酸丁酯偶联制备化合物7r:25ml反应瓶中,称入2mol%醋酸钯与4mol%二茂铁基嘧啶类多齿配体6h,加入12水,搅拌5min后,依次加入4mmol对硝基氯苯、4.8mmol丙烯酸丁酯、6mmol磷酸钾和0.8mmol四丁基溴化铵,升温至80℃反应至反应完全(约6h后反应无继续进行趋势,反应瓶中有钯黑出现),加入25ml乙酸乙酯,水洗三次,合并有机相,无水硫酸钠干燥,浓缩,剩余物通过柱色谱分离,pe/ea=12:1为洗脱剂,得黄色固体 707mg,收率71%。1hnmr(300mhz,cdcl3)δ8.22(d,j=8.7hz,2h), 7.64-7.71(m,3h),6.54(d,j=15.9hz,1h),4.21(t,j=6.6hz,2h), 1.65-1.70(m,2h),1.38-1.46(m,2h),0.95(t,j=7.2hz,3h)ppm;
13
cnmr (75mhz,cdcl3)δ166.1,148.4,141.6,140.6,128.6,124.1,122.6, 65.0,30.7,19.1,13.7ppm。
[0052]
反应条件:卤代苯(1mmol),烯烃(1.2mmol),醋酸钯(0.5mol%),配体6h(1mol%),k3po4(1.5mmol),tbab(0.2mmol)和水(3ml),40-80℃条件下空气中反应,产率为分离提纯后收率。
[0053]
从表3中可以看出,该催化体系具有良好的底物普适性。当卤代烃为碘苯时,反应
能够在较低温度和较短时间内完成(40℃,2-3h)(实验1-13)。碘苯上取代基的位置对反应几乎没有影响(实验6-7)。相反,电子效应对反应的影响较大:当卤代烃上有硝基、酯基和羧基等吸电子基时,反应效果更好(比较实验 7-10与11-14)。这可归因于heck偶联反应中钯对c-x键的插入是整个反应的决速步,吸电子基团的存在使c-x键更容易断裂。当卤代烃为溴苯时,反应仍然可以在较温和的条件进行并且以理想的收率生成偶联产物(实验15和16)。即使是不活泼的氯代烃,稍微升高反应温度和适当延长反应时间,反应也能够顺利进行(实验17和18)。这说明发展的催化体系具有令人满意的催化活性。值得一提的时,该催化体系对一些含有敏感基团的底物同样适用,如甲酰基、羟基和羧基(实验8,10和13)。
[0054]
以上显示和描述了本发明的基本原理和主要特征以及本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
