一种具有抗肿瘤活性齐墩果酸a环衍生物及其制备方法
技术领域
1.本发明涉及一种天然齐墩果酸结构修饰物及制备方法,特别是涉及一种具有抗肿瘤活性齐墩果酸a环衍生物及其制备方法。
背景技术:
2.齐墩果酸 (oleanolic acid , oa),又名庆四素,为齐墩果烷型五环三萜类化合物,广泛分布于植物界中,如在青叶胆全草以及女贞果实等植物中以游离的形式或与糖结合成苷的形式存在。研究表明,齐墩果酸具有多种生物活性,如:降糖、降酯、抗肿瘤、消炎以及抑制血小板聚集等。在临床上作为治疗肝炎的理想药物,毒性低且副作用小,被应用于肝炎的辅助治疗。
3.齐墩果酸的化学结构式为:
技术实现要素:
4.本发明的目的在于提供一种具有抗肿瘤活性的齐墩果酸a环衍生物及其制备方法,该方法以齐墩果酸为先导化合物,设计合成一系列具有抗肿瘤活性的齐墩果酸a环衍生物,该类化合物对人胃癌细胞(sgc-7901)、人肺癌细胞(a549)、肝癌细胞(hepg2)以及口腔癌细胞(kb)具有较强的抑制活性。
5.本发明的目的是通过以下技术方案实现的:一种具有抗肿瘤活性的齐墩果酸a环衍生物,对齐墩果酸a环和c-28位羧基进行结构修饰,分别得到齐墩果酸a环骈合吡嗪环、取代吡嗪环、咪唑环和氢化吡嗪环的c-28位羧酸酯类或酰胺类齐墩果酸衍生物。所述齐墩果酸衍生物包括以下四类:
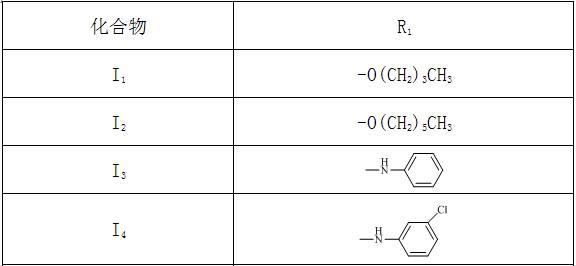
1)在齐墩果酸结构基础上进行修饰,将a环骈合吡嗪环,同时c-28位羧基与卤代烷或胺反应生成酯或酰胺得到一系列化合物。其结构如下表所示:或胺反应生成酯或酰胺得到一系列化合物。其结构如下表所示:2)在齐墩果酸结构基础上进行修饰,将a环骈合取代吡嗪环,同时c-28位羧基与胺反应生成酰胺得到一系列化合物。其结构如下表所示:
3)在齐墩果酸结构基础上进行修饰,将a环骈合咪唑环,同时c-28位羧基与卤代烷反应生成酯得到一系列化合物。其结构如下表所示:合物。其结构如下表所示:4)在齐墩果酸结构基础上进行修饰,将a环骈合氢化吡嗪环,同时c-28位羧基与胺反应生成酰胺得到一系列化合物。其结构如下表所示:
12-烯-28-酰胺类化合物(oa-9-1~oa-9-5)。
[0018] (14)oa-9-1~oa-9-5按照(1)的方法,生成2,3-二羰基-齐墩果-12-烯-28-酰胺类化合物(oa-10-1~oa-10-5)。
[0019]
(15)oa-10-1~oa-10-5分别与乙二胺反应,将a环环合成氢化吡嗪环,生成2,3-并氢化吡嗪环-齐墩果烷型-12-烯-28-酰胺类目标化合物ⅳ1
~ⅳ5
。
[0020]
本发明的优点与效果是: 本发明对五环三萜类天然产物齐墩果酸结构进行化学修饰及改造,得到一系列齐墩果酸结构衍生物。经过药理实验证明,它们分别对人胃癌细胞(sgc-7901)、人肺癌细胞(a549)、肝癌细胞(hepg2)以及人口腔癌细胞(kb)具有较强的抑制作用,且优于母体化合物齐墩果酸。
[0021]
具体实施方法下面结合实施例对本发明做进一步详述。
[0022] 1)齐墩果酸的c-3位羟基被jones试剂氧化,生成3-羰基-齐墩果烷型-12-烯-28-羧酸(oa-1),,在叔丁醇中与叔丁醇钾反应将c-2位氧化成羰基,生成2,3-二羰基-齐墩果烷型-12-烯-28-羧酸(oa-2),中间体(oa-2)与乙二胺、氢氧化钾反应将a环环合成吡嗪环,最后c-28位的羧基与相应的卤代烷或胺反应成酯或酰胺得到2个2,3-齐墩果烷型-12-烯-28-羧酸酯类化合物ⅰ1
~ⅰ2
和5个2,3-齐墩果烷型-12-烯-28-羧酰胺类化合物ⅰ3
~ⅰ7
。
[0023]
其中:r1为
ꢀ‑
o(ch2)3ch3、-o(ch2)5ch3、、、、、。
[0024]
2)oa-2与1,2-丙二胺反应,将a环环合成取代吡嗪环,生成5'-甲基-齐墩果-2-烯并[2,3-b]吡嗪-12-烯-28-羧酸(oa-5),中间体(oa-5)与草酰氯反应,进一步与相应的胺反应将c-28位羧基成酰胺得到5个5'-甲基-齐墩果-2-烯并[2,3-b]吡嗪-12-烯-28-酰胺类化合物ⅱ1
~ⅱ5
。
[0025]
其中:r2为、、、、。
[0026]
3)oa-1与相应的卤代烷和碳酸钾在碱性条件下反应,生成3-羰基-齐墩果-12-烯-28-羧酸酯类化合物(oa-6),进一步与水合肼反应,生成2-腙基-齐墩果-12烯-28-羧酸酯类化合物(oa-7),中间体(oa-7)与甲酸加热回流,使a环环合为咪唑环得到5个齐墩果-2-烯并[2,3-b]咪唑-12-烯-28-羧酸酯类化合物ⅲ1
~ⅲ5
。
[0027]
其中:r3为
ꢀ‑
ch2ch3、-(ch2)2ch3、-(ch2)3ch3、-(ch2)3ch3、-(ch2)5ch3。
[0028] 4)oa-1与草酰氯反应,进一步与相应的胺反应生成3-羰基-12-烯-齐墩果烷-28-酰胺类化合物(oa-8),中间体(oa-8)与间氯过氧苯甲酸反应,生成2-羟基-3-羰基-齐墩果-12-烯-28-酰胺类化合物(oa-9),中间体(oa-9)与jones试剂反应,将c-2位羟基氧化成羰基得到2,3-二羰基-齐墩果-12-烯-28-酰胺类化合物(oa-10),中间体(oa-10)与乙二胺反应,得到5个2,3-并氢化吡嗪环-齐墩果烷型-12-烯-28-酰胺类目标化合物ⅳ1
~ⅳ5
。
[0029]
其中:r4为、、、、-c4h9。
[0030]
以吉非替尼和依托泊苷为阳性对照,采取mtt法对齐墩果酸及其所合成i类化合物进行初步的体外抗肿瘤活性测试。研究表明,部分化合物对人胃癌细胞(sgc-7901)和人肺癌细胞(a549)具有较强的抑制作用,且强于齐墩果酸母体。化合物结构以及体外实验结果如下表:如下表:注:a.化合物浓度在10-5
mol/l时测得的抑制率;b.ic
50
表示半数有效抑制浓度。
[0031] 以人胃癌细胞(sgc-7901)和人肺癌细胞(a549)为靶细胞,采取mtt法对合成的齐墩果酸衍生物进行初步的体外抗肿瘤活性测试,结果表明,齐墩果酸衍生物对sgc-7901细胞和a549细胞表现出较强的抑制作用,且优于母体化合物齐墩果酸。其中i5和
ꢀⅱ1对sgc-7901和a549细胞的抑制作用均较强于阳性对照药物吉非替尼和依托泊苷,表现出较强的抑制作用,这说明将齐墩果酸a环引入吡嗪环,同时c-28位羧基成酯或酰胺可以明显地提高齐墩果酸抗肿瘤活性。
[0032]
以吉非替尼和阿霉素为阳性对照,采取mtt法对齐墩果酸及其所合成ⅱ类和ⅲ类
化合物进行初步的体外抗肿瘤活性检测。研究表明所合成的化合物对人胃癌细胞(sgc-7901)和人肝癌细胞(hepg2)具有较强的抑制作用,化合物结构以及体外实验结果如下表:7901)和人肝癌细胞(hepg2)具有较强的抑制作用,化合物结构以及体外实验结果如下表:注:a.化合物浓度在10-5
mol/l时测得的抑制率;b.ic
50
表示半数有效抑制浓度。
[0033]
以人胃癌细胞(sgc-7901)和人肝癌细胞(hepg2)为靶细胞,采取mtt法对合成的齐墩果酸衍生物进行初步的生物活性测试,结果表明,齐墩果酸衍生物对sgc-7901细胞和hepg2细胞表现出较强的抑制作用,且优于母体化合物齐墩果酸。其中ⅱ5
和ⅲ4
对sgc-7901和hepg2细胞的抑制作用均强于阳性对照药物吉非替尼和阿霉素,表现出较强的抑制作用,这说明将齐墩果酸a环引入取代吡嗪环和咪唑环,同时在c-28位羧基成酯或酰胺可以明显地提高齐墩果酸抗肿瘤活性。
[0034]
以5-氟尿嘧啶和阿霉素为阳性对照,采取mtt法对齐墩果酸及其所合成ⅳ类化合
2。
[0038]
将中间体oa-2 (0.5g,0.001mol),溶于乙醇5ml,加入硫酸镁0.7g,缓慢滴加乙二胺-乙醇溶液(1滴乙二胺),室温反应6h,tlc监测反应终点,加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水mgso4干燥,过滤,减压回收溶剂得淡黄色油状物oa-3。
[0039]
将中间体oa-3(0.50g,0.001mol)溶于无水乙醇20ml,加入氢氧化钾(0.07g,0.001mol),二氧化锰(0.26g,0.003mol),80oc水浴回流8 h,tlc监测反应终点。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水mgso4干燥,过滤,减压回收溶剂得黄色油状物oa-4。
[0040] 将中间体oa-4 (0.50g,0.001mmol)溶于n,n-二甲基甲酰胺12ml中,缓慢滴加溴乙烷12滴(0.005mol),无水碳酸钾(0.30g,0.002mol),室温反应5h,tlc监测反应终点。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂,粗品采用干法上柱,经硅胶柱色谱纯化,洗脱剂为石油醚/乙酸乙酯=15/1(v/v),得白色粉末状固体,产率43.20%。mp 134.3~138.1℃。esi-ms:518.3 (m h) ;1h-nmr(cdcl3,300mhz) δ: 8.27,8.41 (m, 2h, nchchn), 2.87 (m, 1h, h-3), 5.29~5.38 (m, 1h, h-12), 2.50 (t, j = 7.0 hz, 1h, h-18), 4.12 (m, 2h, cooch2ch3), 1.93 (t, j = 11.7 hz, 3h, cooch2ch3), 1.76, 1.68, 1.47 (s, 9h, ch3
×
3), 1.20, 1.12 (s, 6h, ch3
×
2), 1.07 (t, j = 7.5 hz, 1h, h-9), 0.95 (s, 3h, ch3), 0.92 (t, j = 6.5 hz, 1h, h-5), 0.81 (s, 3h, ch3)。
[0041]
实施例2齐墩果-2-烯并[2,3-b]咪唑-12-烯-28-羧酸乙酯(ⅲ1
)的制备 将齐墩果酸oa(0.500g)溶于50ml丙酮中,冰浴下缓慢滴加jones试剂1ml,温度升至室温继续反应1.5 h,tlc监测反应终点,加入15 ml异丙醇淬灭反应。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂,粗品经甲醇重结晶纯化得0.490g 白色粉末状固体oa-1。
[0042]
将中间体oa-1(0.500g),溶于n,n-二甲基甲酰胺(dmf)12ml中,加入无水碳酸钾0.30g,缓慢滴加溴乙烷12滴(0.24ml,5.02mmol),室温反应5h,tlc检测反应终点。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,粗品采用干法上柱,经硅胶柱色谱纯化(洗脱剂:石油醚/乙酸乙酯=15/1(v/v)),得0.324g白色片状固体oa-6。
[0043]
将中间体oa-6 (0.300g),溶于30ml甲醇中,加入水合肼1ml,升温至70℃回流反应2h,tlc检测反应终点。减压回收溶剂,加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,粗品采用干法上柱,经硅胶柱色谱纯化(洗脱剂:石油醚/乙酸乙酯=20/1(v/v))得0.226g黄色油状物oa-7。
[0044]
将中间体oa-7(0.001mol) 溶于20ml无水乙醇中,加入甲酸(0.001mol)升温至70℃回流反应3h,tlc检测反应终点。向反应液中加入50ml冰水静置析晶,抽滤得滤饼,干燥。粗品采用干法上柱,经硅胶柱色谱纯化(洗脱剂:石油醚/乙酸乙酯=20/1(v/v)),得白色晶体iii1,产率63.29%. mp 196.2~199.7 o
c. 1
h-nmr(cdcl3,300mhz) δ:7.52(s,1h,nchn),5.24(t,j=3.2hz,1h, h-12),2.63(t, j=6.5hz,1h,h-18),4.22~4.06(m,2h,cooch2ch3),
1.21(t,j=11.3hz ,3h,cooch2ch3),1.18(s,3h),0.99(s,3h),0.99(s,3h),1.01(s,3h),0.99(s,3h),0.89(s,3h),0.89(s,3h) ;
13
c-nmr(cdcl
3,
150mhz)δ:178.9,165.5,155.3,143.2,123.1,62.3,53.7,51.6,47.6,47.0,46.9,42.5,42.1,40.5,39.5,37.9,34.8,33.7,33.1,32.8,30.9,29.4,27.5,27.5,26.6,25.0,23.8,23.6,23.6,21.0,17.3,15.9,14.1; esi-ms(m/z):506.4[m h]
. elemental anal.(%) calcd. for c
33h50
n2o2: c 78.21,h 9.95,n 5.53,o 6.31;found: c 78.24,h 9.97,n 5.49,o 6.29。
[0045]
实施例3 5',6'-二氢-齐墩果-2-烯并[2,3-b]吡嗪-12-烯-28-酰-3''-氟-4''-氯苯胺(ⅳ1
)的制备将oa(0.100g,0.22mmol)溶于10ml丙酮,冰浴缓慢滴加jones试剂3滴(0.08ml),室温反应1h,tlc监测反应终点,加入异丙醇3ml淬灭。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂,粗品采用干法上柱,经硅胶柱色谱纯化[洗脱剂:v(石油醚)/v(乙酸乙酯)=6/1],得0.095g白色粉末状固体oa-1。
[0046]
将中间体oa-1(0.100g,0.22mmol),溶于5ml二氯甲烷(dcm)中,加入草酰氯(0.24mmol),室温搅拌20h,生成3-氧代-12-烯-齐墩果烷-28-酰氯,旋蒸除去反应溶剂和未反应的草酰氯。酰氯中加入3ml二氯甲烷,加三乙胺调ph为9~10,搅拌5min后,加入3-氟-4-氯-苯胺(0.44mmol),室温反应6h,tlc检测反应终点。反应结束后,向反应液中加入50ml二氯甲烷稀释,用稀盐酸洗涤(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂。粗品采用干法上柱,经硅胶柱色谱纯化[洗脱剂:v(石油醚)/v(乙酸乙酯)=8/1],得0.123g白色晶体oa-8。
[0047]
将中间体oa-8(0.164g,0.3mmol)溶于5ml二氯甲烷中,加入10ml1.5%的硫酸-甲醇溶液,间氯过氧苯甲酸(m-cpba)(0.06g,0.3mmol),冰浴反应24h,tlc检测反应终点。加入na2so3饱和溶液淬灭0.5h,二氯甲烷稀释,用稀盐酸洗涤(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂得0.139g黄色油状液体oa-9。
[0048]
将中间体oa-9,按照oa-1的合成方法制备, 得到 0.121 g 浅黄色固体oa-10。
[0049]
将中间体oa-10(0.168g,0.3mmol)溶于10ml无水乙醇中,加入无水硫酸镁0.150g,缓慢滴加饱和乙二胺-乙醇溶液(2滴,乙二胺,0.6mmol),加热回流5h,tlc检测反应终点。加入乙酸乙酯稀释,饱和食盐水洗涤有机层(100ml
×
3)合并有机相。无水na2so4干燥,过滤,减压回收溶剂,粗品采用干法上柱,经硅胶柱色谱纯化[洗脱剂:v(石油醚)/v(乙酸乙酯)=15/1],得0.096g白色粉末状固体ⅳ1
,产率35.78%.m.p.163.1~165.2℃;1h-nmr(cdcl3,500mhz)δ:7.99(s,1h,nh),7.34(t,j=9.1hz,1h),7.32(s,1h),7.26(s,1h),5.28(s,1h),2.36(t,j=7.5hz,1h),1.56~1.51,1.49~1.42(m,4h,nch2ch2n),1.57~1.40(m,6h),1.47(t,j=7.0hz,1h),1.39~1.37,1.36~1.34(m,4h,nch2ch2n),1.35~1.33(m,4h),1.31~1.24(m,4h),1.23(s,3h),1.18(s,3h),1.14(s,3h),1.12(s,3h),1.02(s,3h),0.96(s,3h),0.95(s,3h);13cnmr(150mhz,cdcl3)δ:180.4,165.9,165.5,163.2,150.6,141.3,131.9,123.4,118.8,117.2,109.2,51.9,51.2,50.2,48.6,45.3,43.7,40.1,36.8,35.7,34.5,34.3,31.2,30.5,30.1,29.8,28.4,28.1,25.3,24.6,24.5,24.2,22.2,20.1,20.1,18.9,18.3,18.1;esi-ms m/z:619.3[m+h]
+
.anal.calcd for c
38h51
clfn3o:c 73.58,h 8.29,n 6.77;found c 73.50,h 8.18,n 6.65。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
