一种氧杂降冰片烯环上c-5位炔基取代的斑蝥素衍生物及其制备方法与应用
技术领域
1.本发明属于化学合成技术领域,具体涉及一种氧杂降冰片烯环上c-5位炔基取代的斑蝥素衍生物及其制备方法与应用。
背景技术:
2.炎症是临床常见的一种病理过程,可发生于机体各组织和器官。炎症免疫反应是机体炎症免疫相关组织、器官和细胞依据内外环境变化所表现出的适度或异常的系统反应,几乎涉及机体绝大多数系统的保护或病理反应。机体炎症免疫反应的异常会表现为炎症性疾病或自身免疫性疾病等,严重影响生物体机能的发挥。根据发病时间,炎症性疾病可分为急性炎症性疾病和慢性炎症性疾病。急性炎症是身体对抗外来病原体入侵和组织损伤的正常反应,但是当体内促炎因子和抑炎因子失衡时,炎症会持续性存在而表现为慢性炎症,引起多种器官和组织的病变,进而会引发包括心血管疾病、脑部疾病、呼吸道疾病、消化道疾病、甚至癌症在内的其他更为严重的疾病。
3.抗炎药物是用于治疗组织受到损伤后所致炎症反应的药物,临床上广泛用于炎症性疾病和自身免疫性疾病等的治疗。目前临床上常用的抗炎药物主要有两大类,以糖皮质激素为代表的甾体抗炎药和以阿司匹林为代表的非甾体抗炎药(或称解热镇痛抗炎)。甾体抗炎药虽然具有强大的抗炎作用,但可能产生激素依赖、免疫抑制、消化系统及代谢系统等严重不良反应;非甾体抗炎药虽然种类繁多,但大多存在副作用大、缓解率低、复发率高等缺陷。为了达到更佳的治疗效果和降低药物不良反应的目的,新型抗炎药物的研发一直是药物研发领域的研究热点。
4.恶性肿瘤是严重威胁人类健康的常见病和多发病,其发病率逐年上升,是导致人类死亡的主要病因之一。目前针对恶性肿瘤的治疗手段主要包括手术、放疗和药物治疗。细胞毒性抗肿瘤药物是传统肿瘤治疗药物中的主力军,是能够直接杀伤肿瘤细胞或抑制肿瘤细胞生长、增殖的一类化疗药物,是治疗恶性肿瘤的主要手段之一,如紫杉醇(paclitaxel)、喜树碱(camptothecin)、长春碱(vinblastine)和鬼臼毒素(podophyllotoxin)及其衍生物等皆属于细胞毒性抗肿瘤药物,它们在临床上被广泛用于各种癌症的治疗并取得了较好的治疗效果。
5.斑蝥素提取自传统中药斑蝥,是中药斑蝥的主要抗癌活性成分,但因毒性较大,从而限制了其临床应用。去甲斑蝥素作为斑蝥素的一种标志性衍生物,是斑蝥素经水解去1,2位甲基后的产物,相比于斑蝥素,去甲斑蝥素不仅保留了斑蝥素的抗癌效果,而且明显降低了斑蝥素对泌尿系统的副作用,并具有一定的免疫调节作用。但去甲斑蝥素具有靶向性差,在血液中的半衰期短的缺点,同样极大地限制了其在临床上的应用。因此基于降毒增效目的的斑蝥素衍生物合成一直是新药研发的热点之一。目前文献报道的关于斑蝥素衍生物的合成改造大多集中在对其结构中丁二酸酐环上的改造,而对其氧杂降冰片烯环上c-5(c-5')和c-6(c-6')位,由于合成方法所限,对其修饰改造的研究还报道较少。
6.本发明旨在提供一种具有肿瘤细胞毒活性和抗炎活性的氧杂降冰片烯环上c-5位修饰的新型斑蝥素衍生物性。
技术实现要素:
7.本发明的第一目的在于提供一种氧杂降冰片烯环上c-5位炔基取代的斑蝥素衍生物及其药学上可接受的盐,本发明的第二目的在于提供所述斑蝥素衍生物的制备方法,本发明的第三目的在于提供所述斑蝥素衍生物的应用。
8.本发明的第一目的是这样实现的,氧杂降冰片烯环上c-5位炔基取代的斑蝥素衍生物及其药学上可接受的盐,其结构通式如式(i)所示:;其中,r为r1或r2;r1选自4-甲基苯乙炔基、2-甲氧基苯乙炔基、3,5-二甲氧基苯乙炔基、4-氟苯乙炔基、4-氯苯乙炔基、4-溴苯乙炔基或三甲基硅乙炔基;r2为乙炔基。
9.所述斑蝥素衍生物化合物优选为:dimethyl 5-((trimethylsilyl)ethynyl)-7-oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylat(化合物1);dimethyl 5-((2-methoxyphenyl)ethynyl)-7
‑ꢀ
oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物2);dimethyl 5-ethynyl-7-oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物3);dimethyl 5-(phenylethynyl)-7-oxabicyclo [2.2.1]hept-2-ene-2,3-dicarboxylate(化合物4);dimethyl 5-(p-tolylethynyl)-7-oxabicyclo [2.2.1]hept-2-ene-2,3-dicarboxylate(化合物5);dmethyl 5-((3,5-dimethoxyphenyl)ethynyl)-7
ꢀ‑
oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物6);dimethyl 5-((4-fluorophenyl)ethynyl)-7
‑ꢀ
oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物7);dimethyl 5-((4-chlorophenyl)ethynyl)-7
ꢀ‑
oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物8);dimethyl 5-((4-bromophenyl)ethynyl)-7
‑ꢀ
oxabicyclo[2.2.1]hept-2-ene-2,3-dicarboxylate(化合物9);化合物1-9的结构式分别如式(iii)中1-9所示。
[0010]
本发明的第二目的是这样实现的,所述斑蝥素衍生物的制备方法,按以下步骤实现:1)以呋喃和丁炔二酸二甲酯为底物在70-90℃下反应7-9h后,经硅胶柱层析,得到结构式如式(ii)所示的中间体a;;2)将[ir(cod)cl]2和(
±
)-binap溶于1,2-二氯乙烷中,在室温下搅拌反应25-35分钟后加入所述中间体a,然后再加入 1,2-二氯乙烷搅拌15-20分钟得到反应液b;3)在反应液b中加入带取代基的端炔化合物,密封后在60-80℃的油浴锅中加热直至底物反应完全,得到反应液c;4)将步骤3中所述反应液c冷却至室温,经浓缩以及硅胶柱层析纯化,得到目标化合物。
[0011]
所述斑蝥素衍生物的应用具体为在制备体外抗肿瘤药物或体内外抗炎药物中的应用。
[0012]
本发明的有益效果为:1、本发明提供了9个结构新颖的斑蝥素衍生物化合物,该化合物1-9具有较优的体内和体外抗炎活性,对于体外抗炎活性,本发明化合物1-9较对照品斑蝥素的选择指数(si)更高,说明本发明化合物1-9的体外抗炎活性并非完全由于其细胞毒性引起,即具有较低的
细胞毒性。另外,本发明化合物1-9还具有优良的抗肿瘤活性,其中有4种化合物的活性在四种肿瘤细胞上较对照品斑蝥素活性强。从而证明了本发明斑蝥素衍生物在抗炎药物及抗肿瘤药物的开发中具有很好的前景。
[0013]
2、本发明先利用丁炔二酸二甲酯和呋喃合成了中间体a,再以a为底物进行结构修饰,得到其他一系列的斑蝥素衍生物。该方法的反应条件温和,合成过程中无有害物质产生,绿色环保,产率高,成本低,操作方便,既可小剂量反应,也适用于工业生产。
附图说明
[0014]
图1为化合物1的1h nmr谱图;图2为化合物1的
13
c nmr谱图;图3为化合物2的1h nmr谱图;图4为化合物2的
13
c nmr谱图;图5为化合物5的1h nmr谱图;图6为化合物5的
13
c nmr谱图;图7为化合物7的1h nmr谱图;图8为化合物7的
13
c nmr谱图;图9为化合物9的1h nmr谱图;图10为化合物9的
13
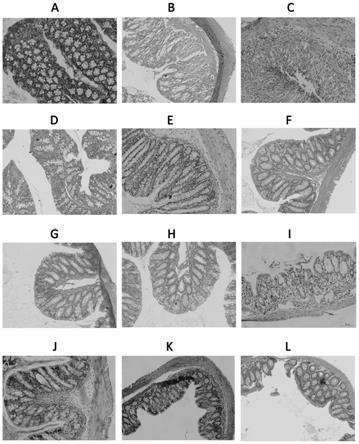
c nmr谱图;图11为化合物 1-9对dss所致小鼠结肠炎的治疗效果的h&e染色图,其中,a为溶媒对照组h&e染色图;b和c为dss模型组h&e染色图;d-l为化合物1-9给药组h&e染色图。
具体实施方式
[0015]
下面结合实施例对本发明作进一步的说明,但不以任何方式对本发明加以限制,基于本发明教导所作的任何变换或替换,均属于本发明的保护范围。
[0016]
本发明提供了一种氧杂降冰片烯环上c-5位炔基取代的斑蝥素衍生物及其药学上可接受的盐,其结构通式如式(i)所示:;其中,r为r1或r2;r1选自4-甲基苯乙炔基、2-甲氧基苯乙炔基、3,5-二甲氧基苯乙炔基、4-氟苯乙炔基、4-氯苯乙炔基、4-溴苯乙炔基或三甲基硅乙炔基;r2为乙炔基。
[0017]
本发明还提供了所述斑蝥素衍生物的制备方法,按以下步骤实现:1)以呋喃和丁炔二酸二甲酯为底物在70-90℃下反应7-9h后,经硅胶柱层析,得到结构式如式(ii)所示的中间体a;
;2)将[ir(cod)cl]2和(
±
)-binap溶于1,2-二氯乙烷中,在室温下搅拌反应25-35分钟后加入所述中间体a,然后再加入1,2-二氯乙烷搅拌15-20分钟得到反应物b;3)在反应物b中加入带取代基的端炔化合物,密封后在60-80℃的油浴锅中加热直至底物反应完全,得到反应液c;4)将步骤3中所述反应液c冷却至室温,经浓缩以及硅胶柱层析纯化,得到目标化合物。
[0018]
当r为r2时,斑蝥素衍生物的制备方法,按以下步骤实现:1)以呋喃和丁炔二酸二甲酯为底物在70-90℃下反应7-9h后,经硅胶柱层析分离纯化,得到结构式如式(ii)所示的中间体a;;2)将[ir(cod)cl]2和(
±
)-binap溶于1,2-二氯乙烷中,在室温下搅拌反应25-35分钟后加入所述中间体a,然后再加入1,2-二氯乙烷搅拌15-20分钟得到反应物b;3)在反应物b中加入三甲基乙炔基硅烷,密封后在60-80℃的油浴锅中加热直至底物反应完全,得到反应液d;4)将步骤3中所述反应液d冷却至室温,经浓缩以及硅胶柱层析纯化后,加入四氢呋喃,于0℃下滴入四丁基氟化铵至反应完全,再经硅胶柱层析分离纯化,得到目标化合物。
[0019]
步骤3中,所述带取代基的端炔化合物为三甲基乙炔基硅烷、2-甲基苯乙炔、苯乙炔、4-甲基苯乙炔、3,5-二甲基苯乙炔、4-氟苯乙炔、4-氯苯乙炔或4-溴苯乙炔中的任意一种。
[0020]
所述步骤1中,呋喃与丁炔二酸二甲酯的摩尔质量比为1:1.5。
[0021]
加入的[ir(cod)cl]2、(
±
)-binap和中间体a的摩尔质量比为0.025:0.065:1。
[0022]
带取代基的端炔化合物与中间体a的摩尔质量比为2:1。
[0023]
本发明斑蝥素衍生物或其药学上可接受的盐的应用为在制备用于体外或体内抗炎药物中的应用。
[0024]
本发明斑蝥素衍生物或其药学上可接受的盐在的另一应用为在制备抗癌药物中的应用。
[0025]
所述斑蝥素衍生物或其药理学上容许的盐,可以列举例如与盐酸、硝酸、硫酸、磷
236.2230。
[0033]
反应式如下:实施例5化合物4的合成在手套箱中,氩气氛围下,将[ir(cod)cl]2(3.4 mg, 0.005 mmol)和(
±
)-binap (8.6 mg, 0.013 mmol)溶于1 ml 1,2-二氯乙烷中,室温下搅拌络合30分钟后,加入实施例1制备的中间体a(42.04mg, 0.2 mmol),然后再加入1 ml 1,2-二氯乙烷,搅拌20分钟后,加入苯乙炔(28.6mg, 0.28 mmol),封口后至于70 ℃的油浴中加热直至底物反应完全(tlc检测)。反应液冷却至室温后,经过浓缩和硅胶柱层析纯化,得化合物4,产率为30%。dimethyl 5-(phenylethynyl)-7-oxabicyclo [2.2.1]hept-2-ene-2,3-dicarboxylate (4):淡黄色固体,1h nmr (400 mhz, cdcl3) δ 7.45
ꢀ–ꢀ
7.38 (m, 2h), 7.32
ꢀ–ꢀ
7.25 (m, 3h), 5.40
ꢀ–ꢀ
5.30 (m, 2h), 3.84 (d, j = 5.4 hz, 6h), 2.86 (dd, j = 8.3, 4.0 hz, 1h), 2.18 (ddd, j = 11.9, 4.7, 4.0 hz, 1h), 2.05 (dd, j = 11.8, 8.4 hz, 1h)。hrms calcd for c
32h30
no [m]
: 236.0685, found: 236.0681。
[0034]
反应式如下:= 实施例6化合物5的合成在手套箱中,氩气氛围下,将[ir(cod)cl]2(3.4 mg, 0.005 mmol)和(
±
)-binap (8.6 mg, 0.013 mmol)溶于1 ml 1,2-二氯乙烷中,室温下搅拌络合30分钟后,加入实施例1制备的中间体a (42.04mg, 0.2 mmol),然后再加入1 ml 1,2-二氯乙烷,搅拌20分钟后,加入4-甲基苯乙炔 (91.4mg, 0.28 mmol),封口后至于70 ℃的油浴中加热直至底物反应完全(tlc检测)。反应液冷却至室温后,经过浓缩和硅胶柱层析纯化,得化合物5,产率为80%。dimethyl 5-(p-tolylethynyl)-7-oxabicyclo [2.2.1]hept-2-ene-2,3-dicarboxylate (5): 黄色固体, 1
h nmr (400 mhz, cdcl3)δ 7.34
ꢀ–ꢀ
7.27 (m, 2h), 7.09 (d, j = 7.9 hz, 2h), 5.40
ꢀ–ꢀ
5.28 (m, 2h), 3.83 (d, j = 5.1 hz, 6h), 2.85 (dd, j = 8.3, 4.0 hz, 1h), 2.33 (s, 3h), 2.17 (ddd, j = 11.8, 4.8, 4.0 hz, 1h), 2.03 (dd, j = 11.8, 8.4 hz, 1h),见图5。
13
c nmr (100 mhz, cdcl3) δ 162.56, 162.44, 144.46, 143.23, 142.78, 131.63, 128.19, 127.99, 90.23, 85.80, 85.04, 81.56, 80.78, 52.46, 52.38, 34.10, 30.31 ,见图6。hrms calcd for c
18h16o5 [m]
: 312.3213, found: 312.3210。
162.5, 162.4, 144.5, 142.7, 133.1, 131.5, 122.2, 122.1, 91.5, 85.7, 80.8, 80.6, 52.5, 34.1, 30.4,见图10。 hrms calcd for c
18h15
bro
5 [m] : 390.0098. found:390.0103。
[0039]
反应式如下:试验例1:本发明斑蝥素衍生物化合物1-9的肿瘤细胞毒活性检测本发明化合物1-9对四种肿瘤细胞(人宫颈癌细胞hela,人黑色素瘤细胞a375,人肺癌细胞a549和人乳腺癌细胞mda-mb-231)生长抑制作用的试验结果、试验原理、方法和结果如下:试验原理:药物与细胞共培养后检测肿瘤细胞存活率。磺酰罗丹明b(sulforhodamine b,srb)是一种水溶性蛋白染料,其分子中的磺酸基阴离子,在弱酸性环境下与细胞内蛋白质的碱性氨基酸结合,用碱性溶液溶解细胞内的srb并测其吸光值,由srb 的含量即可得知细胞内的蛋白质含量,并以此代表细胞存活率。
[0040]
试验方法:用含10%胎牛血清的培养液将以上四种肿瘤细胞配制成单细胞悬液并接种于96空细胞培养板,接种密度5
×
103细胞/孔。培养24小时细胞贴壁后,加入不同浓度的待测化合物溶液,继续培养 48 或 72 小时。加入预冷的50%三氯乙酸溶液固定细胞,之后用 srb 溶液染色存活的细胞,最后加入tris溶液溶解srb,并于520nm波长下检测吸光值。记录结果,以浓度为横坐标,细胞存活率为纵坐标绘制曲线,应用reed and muench法计算化合物的 ic
50
值。
[0041]
表1 化合物1-9 对四种肿瘤细胞生长的半数抑制浓度
注:ctd为对照化合物斑蝥素(cantharidin)。
[0042]
试验结果:由表1可知,化合物1-9均具有肿瘤细胞毒活性,大部分化合物的活性在四种肿瘤细胞上与对照品斑蝥素(cantharidin)活性相当。其中,化合物1,化合物3和化合物5对四种肿瘤细胞的抑制活性明显优于照品斑蝥素(cantharidin)。
[0043]
试验例2:本发明化合物1-9的体外抗炎活性检测本发明化合物1-9抑制lps和inf-γ诱导小鼠raw264.7巨噬细胞释放no的实验结果、实验原理、方法和结果如下:试验原理:一氧化氮(no)广泛分布于生物体内各个组织中,作为一种重要的信号分子,具有重要的生物学作用。no的生成受一氧化氮合酶(nos)影响,nos以l-精氨酸(l-arg)为底物,催化其生成l-瓜氨酸并释放no。nos以三种不同的亚型存在于机体之内:正常情况下表达的神经元型一氧化氮合酶(nnos)、内皮型一氧化氮合酶(enos)和损伤后诱导表达的诱导型一氧化氮合酶(inos)。inos主要分布于淋巴细胞中的效应细胞如中性粒细胞、巨噬细胞和单核细胞中,在介导炎症反应中扮演着重要的病理角色。通过lps和inf-γ刺激小鼠raw264.7巨噬细胞,激活并表达inos,inos催化产生大量的no,no在体内或水溶液中极易氧化成no
22-和no
33-,no
33-可通过镉被还原为no
22-。no
22-在碱性条件下和磺胺作用生成重氮化合物,再与n-1-萘基乙二胺盐酸进行偶联反应,该反应生成的产物浓度与no浓度具有线性关系,且在540nm有最大吸收。
[0044]
试验方法:(1)接种细胞:用含10%胎牛血清的dmed培养液将raw264.7细胞配成单细胞悬浮液,以每孔5
×
104个细胞接种到24孔板上。(2)化合物处理:细胞贴壁后,加入不同浓度的待测化合物溶液,2小时后加入10 μg/ml的lps和200 ng/ml的 inf-γ继续培养24小时。(3)测定:取24孔板上清液150 μl,再加入1%磺胺和0.1% n-1-萘基乙二胺盐酸各50 μl,在波长546 nm下测定吸光度。(4)记录结果,以浓度为横坐标,no抑制率为纵坐标绘制量效关系曲线,应用reed and muench法计算化合物的半数抑制浓度(ic
50
)值。同等条件下用srb法检测化合物对raw264.7细胞毒性情况,计算其对raw264.7细胞生长的半数毒性浓度(tc
50
)值。
[0045]
试验结果如表2所示。
[0046]
表2 化合物1-9 对lps诱导小鼠巨噬细胞raw264.7释放no的半数抑制浓度
注:ctd为对照化合物斑蝥素(cantharidin);smt为阳性对照化合物s-甲基异硫脲硫酸盐(s-methylisothiourea sulfate)。
[0047]
结果表明:本发明化合物1-9能够有效地抑制lps导致的小鼠巨噬细胞raw264.7释放no,具有体外抗炎活性。相对于对照样品斑蝥素,本发明化合物1-9其选择指数(si)更高;另外,斑蝥素对lps诱导的小鼠巨噬细胞raw264.7释放no的作用主要由其细胞毒性作用引起,而化合物1-9的体外抗炎活性并非完全由于其细胞毒性引起。
[0048]
试验例3:本发明化合物的体内抗炎活性检测本发明化合物1-9在dss诱导小鼠肠炎模型上的治疗效果的试验结果、试验原理、方法和结果如下:试验原理:葡聚糖硫酸钠(dss)是一种人工合成的硫酸盐多糖,可以作为用来诱导小鼠结肠炎模型的致炎剂。口服葡聚糖硫酸钠可通过破坏肠道内共生菌的分隔,直接损伤结肠上皮细胞,诱发炎症反应。
[0049]
试验方法:(1)分组:将6-8周龄的雌性c57bl/6小鼠分成14组,每组各5只,分别为溶媒对照组,dss模型组,给药1-9组。(2)称重并标记各组c57bl/6小鼠。除溶媒对照组外,所有小鼠饮用4% dss水溶液。同时,给药1-9组小鼠每天分别灌胃给药化合物1-9。(3)7天后处死所有小鼠,并收集小鼠的远端结肠进行h&e染色病理学检测。
[0050]
试验结果:化合物1-9 对dss诱导的小鼠结肠炎的治疗作用如图11所示。其中,图11a为正常饮水的溶媒对照组;图11b和11c为饮用dss水溶液的模型组;图11d-l分别为给药200mg/kg的化合物1-9的给药组。图片放大倍数200
×
。
[0051]
结果显示:从图11可知,dss饮水的模型组小鼠的远端结肠出现充血、水肿,并出现不同程度的结肠溃疡,黏膜水肿、杯状细胞缺失、隐窝肿胀破坏,黏膜和黏膜下层出现不同程度的炎症细胞浸润和上皮细胞损伤(图11b和c)。dss饮水加化合物 1-9治疗的小鼠结肠病理h&e染色图(图11 d-l)可见不同程度的完整细胞形态,细胞结构清晰,炎症呈现不同程度的缓解,图11 d-l的炎症缓解程度依次为:60%、50%、90%、90%、90%、40%、70%、80%和80%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。