1.本发明属于环氧树脂组合物及其应用领域,特别涉及一种环氧树脂组合物及其制备方法和应用。
背景技术:
2.近年来电子电路领域中柔性化、薄型化的需求日益增多,为了保护半导体元件使电路高集中化或者提高连接可靠性,经常使用包含环氧树脂组合物的胶黏剂、密封材料等。特别是包含在高温条件下发生劣化的部件的半导体装置的情况下,其制造工序均需要在低温条件下进行。例如在图像传感器模块的制造工序变为高温时,会导致模块中所使用的透镜等劣化,因此对于制造中所使用的胶黏剂及密封材料要求具有低温固化性。另外,在电子部件的组装和装配中,出于保持可靠性等目的,无论被粘材料的材质为何,经常需要使用耐湿粘结强度可靠性高的胶黏剂及密封剂。进而,还要求使树脂组合物能够使用的时间长,即适用期长。
3.这种电子部件用途的胶黏剂、密封剂中所使用的环氧树脂组合物通常包含环氧树脂和固化剂。其中,所述环氧树脂包含各种多官能环氧树脂(具有2个以上环氧基的环氧树脂),所述固化剂包含具有2个以上能够与环氧树脂中的环氧基反应的官能团的化合物。在此,已知环氧树脂
‑
硫醇固化剂体系的树脂组合物在达成低温快速固化上有效。已知这种树脂组合物中使用硫醇作为固化剂的类型即使在0℃
‑
20℃这一低温条件下也适宜地在短时间内固化。然而,以往的硫醇系固化剂,例如季戊四醇四(3
‑
巯基丙酸酯)(sc有机化学制,商品名:pemp)、三羟甲基丙烷三(3
‑
巯基丙酸酯)(sc有机化学制,商品名:tmmp)及季戊四醇四(3
‑
巯基丁酸酯)(昭和电工制,商品名:karenzmt pe1),具有其固化后的树脂组合物耐湿性差的问题(参见专利jph06211969a和jph06211970a),这是由于以往的硫醇系固化剂在其分子骨架上包含酯键结构。
4.另外,专利us4266055a和jps56120671a中合成的三巯丙基异氰尿酸酯,在分子中不具有酯键,因此被用作耐水性优异的环氧树脂组合物的固化剂,但是该固化剂在室温下会散发出难闻的气味(强烈的硫臭味),固化物的耐热性也不令人满意。此外,为了同时赋予环氧树脂固化物良好的耐湿性及耐热性,目前行业内的主流做法是采用巯基烷基甘脲作为环氧树脂的固化剂。例如,专利cn201480064943.9和jp2015059099a公开了所谓巯基烷基甘脲的多硫醇固化剂,虽然该多硫醇固化剂具有良好的耐湿性及耐热性,但是专利 cn201680014880.5中却提出该多硫醇固化剂在室温下为固体,在与环氧树脂形成配合物时容易析出晶体,存在组成变得不均匀的问题,此时需要与另一种巯基乙基甘脲化合物配合使用,由此使固体多硫醇固化剂液体化,最终形成液体状的具有二硫键的低聚物混合物作为固化剂,虽然该方式能够使固化剂最终转化为液体,但是却增加了反应的工序和成本。再则,cn201480064943.9中提及的巯基烷基甘脲类固化剂还有降低单组份低温固化环氧胶的储存稳定性的风险。
5.总体来看,目前市场上的各类硫醇化合物和含有硫醇化合物的固化剂主要存在以
下问题,导致使用了这些固化剂的环氧树脂组合物总是存在各种缺陷:
6.(1)现有的多硫醇固化剂大多含有酯键,例如季戊四醇四(3
‑
巯基丙酸酯)、三羟甲基丙烷三(3
‑
巯基丙酸酯)以及季戊四醇四(3
‑
巯基丁酸酯),高温高湿环境下容易水解,使用这类酯键型硫醇固化剂的环氧组合物,在高温高湿环境中固化后粘接强度下降明显;
7.(2)现有的多硫醇固化剂,例如无酯键的三巯丙基异氰尿酸酯(专利us4266055a、 jps56120671a),依然会散发出难闻的气味,具有强烈的硫臭味,大多气味很大,从而严重影响施胶时的工作环境;
8.(3)少数无酯键型硫醇,例如1,3,4,6
‑
四(2
‑
巯基乙基)甘脲,虽然可以解决耐湿热、耐热性以及气味的问题,但在室温下为固体,若想变为液态,提升晶体析出时间,需要与一种具有二硫键的巯基乙基甘脲化合物配合使用,因此在制备时需要额外进行偶联形成低聚物混合物,才能变为液态,工艺复杂,成本高。此外,现有的巯基烷基甘脲类固化剂还会提高单组份低温固化环氧胶的储存稳定性的风险,适用期短。
技术实现要素:
9.本发明的第一目的是为了解决上述问题,而提供了一种环氧树脂组合物,其中所使用的硫醇化合物在不含甘脲基的基础上,室温下为液态且无酯键,对应的环氧树脂组合物在储存中无晶体析出,适用期长,具有良好的耐热耐湿性。
10.本发明的第二目的在于提供一种上述环氧树脂组合物的制备方法。
11.本发明的第三目的在于提供一种上述环氧树脂组合物的应用。
12.具体地,本发明提供的环氧树脂组合物中包括以下组分:
13.(a)环氧树脂;
14.(b)下述通式(ⅰ)所示的硫醇化合物;
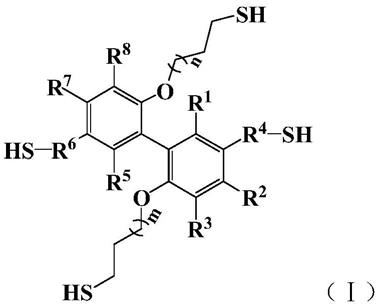
[0015][0016]
所述通式(ⅰ)中,r1、r2、r3、r5、r7和r8分别独立地选自氢原子、碳原子数为 1
‑
5的烷基和碳原子数为1
‑
5的烷氧基中的一种,r4和r6分别独立地选自碳原子数为1
‑
5 的亚烷基,m和n分别独立地为0、1、2或3;以及
[0017]
(c)固化促进剂。
[0018]
在一种优选实施方式中,所述环氧树脂组合物中各组分的含量如下:
[0019]
所述环氧树脂
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
25
‑
65重量份;
[0020]
所述硫醇化合物
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
20
‑
45重量份;
[0021]
所述固化促进剂
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.1
‑
15重量份。
[0022]
在一种优选实施方式中,所述通式(ⅰ)中,r1、r2、r5和r7均为氢原子,r3和r8分别独
立地选自氢原子或甲氧基,r4和r6分别独立地选自碳原子数为3
‑
5的亚烷基,m和 n为1或2。
[0023]
在一种优选实施方式中,所述多硫醇化合物选自5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丁氧基)联苯、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丁氧基)
‑
3,3'
‑
二甲氧基联苯、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基戊氧基)联苯、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基戊氧基)
‑
3,3'
‑
二甲氧基联苯、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯以及5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯中的一种或几种的组合。
[0024]
在一种优选实施方式中,所述硫醇化合物按照包括以下步骤的方法制备得到:
[0025]
步骤一:将由通式(ⅱ)表示的苯酚类化合物和由通式(ⅲ)表示的第一化合物在相转移催化剂的存在下且在碱性条件下进行取代反应,提纯后得到呈液态的无色或淡黄色的第一中间产物;
[0026]
步骤二:将第一中间产物和硫代乙酸在自由基引发剂的存在下进行自由基加成反应,提纯后得到呈液态的无色或淡黄色的第二中间产物;
[0027]
步骤三:将第二中间产物进行水解反应,提纯后得到呈无色或淡黄色的粘稠液体状产物,即为多硫醇化合物;
[0028][0029]
通式(ⅱ)中,r1、r2、r3、r5、r7和r8分别独立地选自氢原子、碳原子数为1
‑
5的低级烷基和碳原子数为1
‑
5的烷氧基中的一种,r9和r
10
分别独立地选自碳原子数为1
‑
5的 1
‑
烯基烷基;
[0030]
通式(ⅲ)中,x表示氯或溴,m为0、1、2或3。
[0031]
在一种优选实施方式中,步骤一中,所述取代反应的方式为将由通式(ⅱ)表示的苯酚类化合物溶解在有机溶剂中,加碱提供碱性条件,加入相转移催化剂,之后在惰性气体保护下升温至40
‑
100℃搅拌10
‑
60分钟,随后再加入通式(ⅲ)表示的第一化合物,反应 4
‑
12小时,接着将反应液过滤,滤液减压蒸馏除去溶剂,用水洗三次,并用三氯甲烷萃取,有机相收集后蒸干,得到呈液态的无色或淡黄色的第一中间产物。
[0032]
在一种优选实施方式中,步骤二中,所述自由基加成反应的方式为将第一中间产物溶解在有机溶剂中,加入自由基引发剂,在惰性气体保护下升温至40
‑
100℃,缓慢加入硫代乙酸,进行自由基加成反应4
‑
12小时,之后减压蒸馏除去溶剂,得到呈液态的无色或淡黄色的第二中间产物。
[0033]
在一种优选实施方式中,步骤三中,所述水解反应的方式为将第二中间产物溶解在有机溶剂中,加入盐酸或氢氧化钠,升温至50
‑
100℃反应3
‑
12小时,减压蒸馏除去溶剂,用 2
‑
8%的碳酸氢钠溶液洗两遍,并用三氯甲烷萃取,有机相收集后蒸干,得到呈无色或淡黄色的粘稠液状产物,即为多硫醇化合物。
[0034]
在一种优选实施方式中,所述环氧树脂为芳香族环氧树脂和/或脂肪族环氧树脂;
当所述环氧树脂同时含有芳香族环氧树脂和脂肪族环氧树脂时,所述脂肪族环氧树脂与芳香族环氧树脂的质量比小于或等于1:4。
[0035]
在一种优选实施方式中,所述环氧树脂的环氧官能团当量与硫醇化合物的硫醇官能团当量之比为0.5
‑
2.0,优选为0.9
‑
1.2。
[0036]
在一种优选实施方式中,所述固化促进剂选自咪唑系固化促进剂、叔胺系固化促进剂和磷化合物系固化促进剂中的至少一种。
[0037]
在一种优选实施方式中,所述环氧树脂组合物中还包括稳定剂;所述稳定剂选自液体硼酸酯化合物、铝螯合剂以及巴比妥酸中的至少一种;所述稳定剂的含量为0.1
‑
5重量份。
[0038]
在一种优选实施方式中,所述环氧树脂组合物中还包括硅烷偶联剂;所述硅烷偶联剂选自3
‑
环氧丙氧基丙基三甲氧基硅烷、2
‑
(3,4
‑
环氧环己基)乙基三甲氧基硅烷、3
‑
甲基丙烯酰氧基丙基三甲氧基硅烷以及8
‑
环氧丙氧基辛基三甲氧基硅烷中的至少一种;所述硅烷偶联剂的含量为0.1
‑
5重量份。
[0039]
在一种优选实施方式中,所述环氧树脂组合物中还包括填料;所述填料选自二氧化硅、氧化铝、氧化镁、氧化锌、氮化硼、碳化硅、滑石、碳酸钙、玻璃微球、石墨粉末、金属粉末以及聚四氟乙烯中的至少一种;所述填料的含量为0.1
‑
40重量份。
[0040]
在一种优选实施方式中,所述环氧树脂组合物中还包括助剂;所述助剂选自阻燃剂、稀释剂、颜料、抗氧化剂、粘接促进剂、消泡剂、流平剂、触变剂、均化剂以及离子捕捉剂中的至少一种;所述助剂的含量为0.1
‑
15重量份。
[0041]
本发明还提供了所述环氧树脂组合物的制备方法,该方法包括将环氧树脂、硫醇化合物、固化促进剂以及任选的稳定剂、硅烷偶联剂、填料和助剂混合均匀即可。
[0042]
本发明还提供了所述环氧树脂组合物在制备胶黏剂中的应用。
[0043]
本发明还提供了所述环氧树脂组合物在制备密封剂中的应用。
[0044]
本发明的有益效果如下:
[0045]
(1)本发明采用具有特定结构的多硫醇化合物作为固化剂,由此能够赋予环氧树脂组合物良好的耐热耐湿性能;
[0046]
(2)本发明采用的多硫醇化合物的气味低,由此能够使环氧树脂组合物避免出现气味过大的情况;
[0047]
(3)本发明采用的多硫醇化合物在常温下为液体,可直接作为固化剂用于树脂组合物的固化,无需额外进行偶联形成低聚物混合物,也无须与其他多硫醇化合物联用,成本低,以该多硫醇化合物作为固化剂的环氧树脂组合物可作为密封剂和粘结剂的成分。此外,本发明采用的多硫醇化合物能够提高单组份低温固化环氧树脂储存稳定性,适用期长,极具工业应用前景。
附图说明
[0048]
图1为5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯的1h
‑
nmr图;
[0049]
图2为5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯的ir光谱图;
[0050]
图3为5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯的1h
‑
nmr图;
[0051]
图4为5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯的ir光谱图。
具体实施方式
[0052]
本发明提供的环氧树脂组合物中包括环氧树脂、通式(ⅰ)所示的硫醇化合物和固化促进剂。其中,所述环氧树脂的含量优选为40
‑
60重量份,例如,可以40、42、45、48、 50、52、55、58、60重量份。所述硫醇化合物的含量优选为20
‑
60重量份,例如,可以为 20、25、30、35、40、45、50、55、60重量份。所述固化促进剂的含量优选为0.5
‑
10重量份,例如,可以为0.5、1、3、5、7、10重量份。
[0053]
所述多硫醇化合物具有通式(ⅰ)所示的结构:
[0054][0055]
所述通式(ⅰ)中,r1、r2、r3、r5、r7和r8分别独立地选自氢原子、碳原子数为 1
‑
5的烷基和碳原子数为1
‑
5的烷氧基中的一种,r4和r6分别独立地选自碳原子数为1
‑
5 的亚烷基,m和n分别独立地为0、1、2或3。其中,所述碳原子数为1
‑
5的烷基的具体实例包括但不限于:甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基、叔戊基或新戊基。所述碳原子数为1
‑
5的烷氧基的具体实例包括但不限于:甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基或异丁氧基。所述碳原子数为1
‑
5的亚烷基的具体实例包括但不限于:亚甲基、亚乙基、正亚丙基、异亚丙基、正亚丁基、仲亚丁基、异亚丁基、叔亚丁基、正亚戊基、异亚戊基、叔亚戊基或新亚戊基。
[0056]
在本发明的一种优选实施方式中,通式(ⅰ)中,r1、r2、r5和r7均为氢原子;r3和r8分别独立地选自氢原子或甲氧基;r4和r6分别独立地选自碳原子数为3
‑
5的亚烷基,如正亚丙基、异亚丙基、正亚丁基、仲亚丁基、异亚丁基、叔亚丁基、正亚戊基、异亚戊基、叔亚戊基或新亚戊基;m和n为1或2。
[0057]
所述多硫醇化合物的具体实例包括但不限于:5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丁氧基) 联苯(r1、r2、r3、r5、r7和r8均为氢原子,r4和r6均为碳原子数为3的亚烷基,m和 n均为2)、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丁氧基)
‑
3,3'
‑
二甲氧基联苯(r1、r2、r5和 r7均为氢原子,r3和r8分别独立地选自甲氧基,r4和r6均为碳原子数为3的亚烷基,m 和n均为2)、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基戊氧基)联苯(r1、r2、r3、r5、r7和r8均为氢原子,r4和r6均为碳原子数为3的亚烷基,m和n均为3)、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑ꢀ
双(3
‑
巯基戊氧基)
‑
3,3'
‑
二甲氧基联苯(r1、r2、r5和r7均为氢原子,r3和r8分别独立地选自甲氧基,r4和r6均为碳原子数为3的亚烷基,m和n均为3)、5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑ꢀ
双(3
‑
巯基丙氧基)
联苯(r1、r2、r3、r5、r7和r8均为氢原子,r4和r6均为碳原子数为3 的亚烷基,m和n均为1)以及5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯(r1、r2、r5和r7均为氢原子,r3和r8分别独立地选自甲氧基,r4和r6均为碳原子数为3的亚烷基,m和n均为1)中的至少一种。
[0058]
在一种具体实施方式中,所述多硫醇化合物按照包括以下步骤的方法制备得到:
[0059]
步骤一:将由通式(ⅱ)表示的苯酚类化合物和由通式(ⅲ)表示的第一化合物在相转移催化剂的存在下且在碱性条件下进行取代反应,提纯后得到呈液态的无色或淡黄色的第一中间产物;
[0060]
步骤二:将第一中间产物和硫代乙酸在自由基引发剂的存在下进行自由基加成反应,提纯后得到呈液态的无色或淡黄色的第二中间产物;
[0061]
步骤三:将第二中间产物进行水解反应,提纯后得到呈无色或淡黄色的粘稠液状产物,即为多硫醇化合物;
[0062][0063]
通式(ⅱ)中,r1、r2、r3、r5、r7和r8分别独立地选自氢原子、碳原子数为1
‑
5的低级烷基和碳原子数为1
‑
5的烷氧基中的一种;优选地,r1、r2、r5和r7均为氢原子,r3和r8分别独立地选自氢原子或甲氧基。r9和r
10
分别独立地选自碳原子数为1
‑
5的1
‑
烯基烷基,优选分别独立地选自碳原子数为3
‑
5的1
‑
烯基烷基。
[0064]
通式(ⅲ)中,x表示氯或溴,m为0、1、2或3。
[0065]
步骤一中,所述取代反应的方式优选为将由通式(ⅱ)表示的苯酚类化合物溶解在有机溶剂中,加碱提供碱性条件,加入相转移催化剂,之后在惰性气体保护下升温至40
‑
100℃搅拌10
‑
60分钟,随后再加入通式(ⅲ)表示的第一化合物,反应4
‑
12小时,接着将反应液过滤,滤液减压蒸馏除去溶剂,用水洗三次,并用三氯甲烷萃取,有机相收集后蒸干,得到呈液态的无色或淡黄色的第一中间产物。
[0066]
所述碱的种类没有特别的限定,可以为领域的常规选择,其具体实例包括但不限于:碳酸钾、碳酸钠、氢氧化钠、氢氧化钾、三乙胺和对二甲氨基吡啶中的至少一种。
[0067]
所述相转移催化剂可以为现有的各种能够催化由通式(ⅱ)表示的苯酚类化合物中的酚羟基与由通式(ⅲ)表示的第一化合物中的氯或溴发生取代反应的物质,优选为环状冠醚类、聚醚类和铵类中的至少一种。其中,所述环状冠醚类的具体实例包括但不限于:18
‑ꢀ
冠
‑
6、15
‑
冠
‑
5和环糊精中的至少一种。所述聚醚类的具体实例包括但不限于:链状聚乙二醇和/或链状聚乙二醇二烷基醚。所述铵类的具体实例包括但不限于:苄基三乙基氯化铵、四丁基溴化铵、四丁基氯化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、十二烷基三甲基氯化铵和十四烷基三甲基氯化铵中的至少一种。
[0068]
步骤二中,所述自由基加成反应的方式优选为将第一中间产物溶解在有机溶剂中,加入自由基引发剂,在惰性气体保护下升温至40
‑
100℃,缓慢加入硫代乙酸,进行自由
基加成反应4
‑
12小时,之后减压蒸馏除去溶剂,得到呈液态的无色或淡黄色的第二中间产物。
[0069]
所述自由基引发剂可以为现有的各种能够引发第一中间产物中的双键与硫代乙酸中的巯基实现自由基加成反应的物质,可以为偶氮类引发剂和/或过氧类引发剂。其中,所述偶氮类引发剂的具体实例包括但不限于:偶氮二异丁腈、2,2'
‑
偶氮双(2
‑
甲基丁腈)、二甲基2,2'
‑ꢀ
偶氮双(2
‑
甲基丙酸酯)、偶氮二异丁酸二甲酯、偶氮二异丁脒盐酸盐、偶氮二甲酰胺、偶氮二异丙基咪唑啉盐酸盐、偶氮异丁氰基甲酰胺、偶氮二环己基甲腈、偶氮二氰基戊酸、偶氮二异丙基咪唑啉、偶氮二异戊腈和偶氮二异庚腈中的至少一种。所述过氧类引发剂的具体实例包括但不限于:叔己基过氧化异丙基单碳酸酯、叔己基过氧化2
‑
乙基己酸酯、1,1,3,3
‑ꢀ
四甲基丁基过氧化2
‑
乙基己酸酯、叔丁基过氧化特戊酸酯、叔己基过氧化特戊酸酯、叔丁基过氧化新癸酸酯、叔己基过氧化新癸酸酯、1,1,3,3
‑
四甲基丁基过氧化新癸酸酯、1,1
‑
双(叔己基过氧化)环己烷、过氧化苯甲酰、3,5,5
‑
三甲基过氧化己酰、过氧化月桂酰和过氧化苯甲酰叔丁酯中的至少一种。从原料易得性的角度考虑,所述自由基引发剂优选为偶氮二异丁腈、2,2'
‑
偶氮双(2
‑
甲基丁腈)、二甲基2,2'
‑
偶氮双(2
‑
甲基丙酸酯)、叔己基过氧化异丙基单碳酸酯、叔己基过氧化2
‑
乙基己酸酯、1,1,3,3
‑
四甲基丁基过氧化2
‑
乙基己酸酯、叔丁基过氧化特戊酸酯、叔己基过氧化特戊酸酯、叔丁基过氧化新癸酸酯、叔己基过氧化新癸酸酯、 1,1,3,3
‑
四甲基丁基过氧化新癸酸酯、1,1
‑
双(叔己基过氧化)环己烷、过氧化苯甲酰、3,5,5
‑ꢀ
三甲基过氧化己酰和过氧化月桂酰中的至少一种。
[0070]
步骤三中,所述水解反应的方式优选为将第二中间产物溶解在有机溶剂中,加入盐酸或氢氧化钠,升温至50
‑
100℃反应3
‑
12小时,减压蒸馏除去溶剂,用2
‑
8%的碳酸氢钠溶液洗两遍,并用三氯甲烷萃取,有机相收集后蒸干,得到呈无色或淡黄色的粘稠液状产物,即为多硫醇化合物。
[0071]
在本发明的一种优选实施方式中,步骤一的取代反应在有机溶剂ⅰ的存在下进行,步骤二的自由基加成反应在有机溶剂ⅱ的存在下进行,步骤三的水解反应在有机溶剂ⅲ的存在下进行。所述有机溶剂ⅰ和有机溶剂ⅱ优选分别独立地选自甲醇、乙醇、丙醇、丁醇、异丙醇、乙酸乙酯、乙酸丙酯、乙酸丁酯、四氢呋喃、二氧杂环己烷、乙腈、甲苯、二甲苯、二氯甲烷、氯仿、四氯化碳、二甲基甲酰胺、二甲基乙酰胺和二甲基亚砜中的至少一种。所述有机溶剂ⅲ优选为醇,更优选为碳原子数为1
‑
5的单元醇,如甲醇、乙醇、丙醇和正丁醇中的至少一种。
[0072]
所述环氧树脂可以为具有两个以上环氧基的脂肪族环氧树脂,也可以为具有两个以上环氧基的芳香族环氧树脂,还可以为两者的混合物。从与硫醇化合物相容性的角度出发,相较于具有两个以上环氧基的脂肪族环氧树脂,所述环氧树脂优选至少含有具有两个以上环氧基的芳香族环氧树脂。其中,所述环氧树脂中所含的具有两个以上环氧基的脂肪族环氧树脂与芳香族环氧树脂的质量比优选小于或等于1:4,例如,可以为0(即仅含有具有两个以上环氧基的芳香族环氧树脂)、1:20、1:15、1:10、1:9、1:8、1:7、1:6、1:5、1:4。
[0073]
所述具有两个以上环氧基的脂肪族环氧树脂可以为二元环氧树脂、三元环氧树脂等中的至少一种。其中,所述二元环氧树脂的具体实例包括但不限于:(聚)乙二醇二缩水甘油醚、(聚)丙二醇二缩水甘油醚、丁二醇二缩水甘油醚、新戊二醇二缩水甘油醚、1,6
‑
己二醇二缩水甘油醚、三羟甲基丙烷二缩水甘油醚、聚四亚甲基醚二醇二缩水甘油醚、甘油二缩
(旭化成公司)、novacure hxa9322hp(旭化成公司)、novacure hxa3922hp(旭化成公司)、novacure hxa3932hp(旭化成公司)、novacure hxa5945hp(旭化成公司)、novacure hxa9382hp(旭化成公司)、fuji cure fxr1121(t&k toka公司) 等,另外,作为脲型加合物系,可列举fuji cure fxe
‑
1000(t&k toka公司)、fuji curefxr
‑
1030(t&k toka公司)等,但不限于这些。上述固化促进剂可以单独使用,也可以将两种以上组合使用。从延长贮存期及改善固化性的角度出发,所述固化促进剂优选为固体分散型胺加合物系潜在性固化促进剂。
[0081]
本发明提供的环氧树脂组合物中,为了提高其储藏稳定性、延长贮存期,优选还添加有稳定剂。所述稳定剂可以为现有的各种以环氧树脂为主剂的单组分型粘接剂的稳定剂,从提高储藏稳定效果的角度出发,所述稳定剂优选选自液体硼酸酯化合物、铝螯合物和巴比妥酸中的至少一种。其中,所述液体硼酸酯化合物的具体实例包括但不限于:2,2
’‑
氧基双(5,5
’‑
二甲基
‑
1,3,2
‑
氧杂己硼烷)、硼酸三甲酯、硼酸三乙酯、硼酸三正丙酯、硼酸三异丙酯、硼酸三正丁酯、硼酸戊酯、硼酸三烯丙酯、硼酸三己酯、硼酸三环己酯、硼酸三辛酯、硼酸三壬酯、硼酸三癸酯硼酸三(十二烷基)酯、硼酸三(十六烷基)酯、硼酸三(十八烷基)酯、硼酸三苯酯、硼酸三邻甲苯酯、硼酸三间甲苯酯、三乙醇胺硼酸酯等中的至少一种。所述液体硼酸酯化合物在常温(25℃)下为液状,因此将配合物粘度抑制为较低,故而优选。所述铝螯合物例如可以为铝螯合物a(川研精密化学公司制)。此外,所述稳定剂的含量优选为0.1
‑
5重量份,例如,0.1、0.5、1、1.5、2、2.5、3、3.5、4、4.5、5重量份。
[0082]
本发明提供的环氧树脂组合物中,可以往环氧树脂组合物中添加硅烷偶联剂。其中,所述硅烷偶联剂可以为环氧系、氨基系、乙烯基系、甲基丙烯酸系、丙烯酸系、巯基系等各种硅烷偶联剂,优选选自3
‑
环氧丙氧基丙基三甲氧基硅烷、2
‑
(3,4
‑
环氧环己基)乙基三甲氧基硅烷、3
‑
甲基丙烯酰氧基丙基三甲氧基硅烷以及8
‑
环氧丙氧基辛基三甲氧基硅烷中的至少一种。此外,所述硅烷偶联剂的含量优选为0.1
‑
5重量份,例如,0.1、0.5、1、1.5、2、 2.5、3、3.5、4、4.5、5重量份。
[0083]
本发明提供的环氧树脂组合物中,可以往环氧树脂组合物中添加填料。在将所述环氧树脂组合物作为胶黏剂或密封剂使用的情况下,若向其中添加填料,则所粘接部位的耐热性、耐湿性、特别是耐热循环性能够得以提高。通过添加填料而使耐热循环性提高的原因在于,固化物的线膨胀系数降低,即由热循环导致的固化物的膨胀
‑
收缩得到抑制。所述填料只要具有降低线膨胀系数的效果即可,没有特别限定,其具体实例包括但不局限于:二氧化硅、氧化铝、氧化镁、氧化锌、氮化硼、碳化硅、滑石、碳酸钙、玻璃微球、石墨粉末、金属粉末以及聚四氟乙烯(ptfe)中的至少一种,优选为二氧化硅和/或氧化铝。此外,所述填料的含量优选为0.1
‑
40重量份,例如,0.1、5、10、15、20、25、30、35、40重量份。
[0084]
根据需要,则可以在不损害本发明主旨的范围内向本发明的环氧树脂组合物中添加其他的助剂,例如阻燃剂、稀释剂、颜料、抗氧化剂、粘接促进剂、消泡剂、流平剂、触变剂、均化剂、离子捕捉剂等中的至少一种。各助剂的种类及其添加量可以为本领域的常规选择。例如,所述助剂的含量可以为0.1
‑
15重量份,例如,0.1、2、5、8、10、12、15重量份。
[0085]
下面将结合制备例、实施例及对比例,对本发明的技术方案进行进一步说明。
[0086]
以下制备例中,在硫醇化合物5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯和5,5'
‑
双 (3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯的制备过程中
用到的原材料来源如下:5,5'
‑
二烯丙基
‑
2,2'
‑
联苯二酚源自萨恩化学技术(上海)有限公司,牌号为e100338;相转移催化剂18
‑
冠
‑
6源自上海泰坦科技股份有限公司,牌号为30243d;烯丙基溴源自上海泰坦科技股份有限公司,牌号为13125c;偶氮二异丁腈(简称“aibn”)源自上海麦克林生化科技有限公司,牌号为a800353;硫代乙酸源自国药集团化学试剂有限公司,牌号为80128126;5,5'
‑
二烯丙基
‑
3,3'
‑
二甲氧基
‑
2,2'
‑
联苯二酚源自萨恩化学技术(上海)有限公司,牌号为d050881。
[0087]
制备例1
[0088]
该制备例用于说明多硫醇化合物(5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯)的制备,具体步骤及反应流程如下:
[0089][0090]
步骤一:将5,5'
‑
二烯丙基
‑
2,2'
‑
联苯二酚80g溶解在200ml丙酮中,加入碳酸钾103.6g 和相转移催化剂18
‑
冠
‑
6 7.9g,在惰性气体保护下升温至70℃搅拌10分钟,然后缓慢加入烯丙基溴79.8g,反应8小时后,将反应液过滤,滤液减压蒸馏除去溶剂,用水洗三次,并用三氯甲烷萃取,有机相收集后蒸干,得到呈液态的淡黄色第一中间产物;
[0091]
步骤二:将步骤二中得到的第一中间产物溶解在200ml四氢呋喃中,加入自由基引发剂偶氮二异丁腈5.4g,在惰性气体保护下升温至70℃,缓慢加入硫代乙酸96.2g,反应12 小时后,减压蒸馏除去溶剂和过量的硫代乙酸,得到呈液态的淡黄色第二中间产物;
[0092]
步骤三:将步骤二中得到的第二中间产物溶解在300ml甲醇中,加入60ml盐酸进行水解,升温至60℃水解反应12小时,减压蒸馏除去溶剂,用5%的碳酸氢钠溶液洗两遍,并用三氯甲烷萃取,有机相收集后蒸干,得到呈淡黄色粘稠液体的最终产物124.8g,即5,5'
‑ꢀ
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯,总产率86.2%,硫醇当量为120g/eq。该5,5'
‑ꢀ
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯基本没有硫臭味。
[0093]
该5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯的1h
‑
nmr图以及ir光谱图
分别如图1和图2所示。从图1可以看出,化学位移7.0ppm附近为联苯环上的质子峰,化学位移4.0ppm、2.6ppm和1.95ppm处的峰对应联苯环上的烷氧基,化学位移2.7ppm、2.5ppm 和1.89ppm处的峰则对应联苯环上的烷基,化学位移1.42ppm和1.3ppm处的峰分别对应联苯环上烷基巯基和烷氧基巯基。从图2可以看出,1492cm
‑1处是联苯环上的吸收峰,815 cm
‑1处对应联苯环上的ar
‑
h弯曲振动,2928cm
‑1处为烷基链上的c
‑
h伸缩振动吸收峰, 1246cm
‑1出现的吸收峰为烷氧基c
‑
o伸缩振动,2560cm
‑1处的吸收峰则对应巯基。由此可以看出,该5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)联苯具有式(ⅳ)所示,其中,r1、 r2、r3、r5、r7和r8均为氢原子,r4和r6均为碳原子数为3的亚烷基,m和n均为1。
[0094][0095]
制备例2
[0096]
该制备例用于说明多硫醇化合物(5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯)的制备,具体步骤及反应流程如下:
[0097]
步骤一:将5,5'
‑
二烯丙基
‑
3,3'
‑
二甲氧基
‑
2,2'
‑
联苯二酚98g溶解在250ml丙酮中,加入碳酸钾103.6g和相转移催化剂18
‑
冠
‑
6 7.9g,在惰性气体保护下升温至70℃搅拌10分钟,然后缓慢加入烯丙基溴79.8g,反应8小时后,将反应液过滤,滤液减压蒸馏除去溶剂,用水洗三次,并用三氯甲烷萃取,有机相收集后蒸干,得到呈液态的淡黄色第一中间产物;
[0098]
步骤二:将步骤一中得到的第一中间产物溶解在200ml四氢呋喃中,加入自由基引发剂偶氮二异丁腈5.4g,在惰性气体保护下升温至70℃,缓慢加入硫代乙酸96.2g,反应12 小时后,减压蒸馏除去溶剂和过量的硫代乙酸,得到呈液态的淡黄色第二中间产物;
[0099]
步骤三:将步骤二中得到的第二中间产物溶解在300ml甲醇中,加入60ml盐酸进行水解,升温至70℃水解反应12小时,减压蒸馏除去溶剂,用5%的碳酸氢钠溶液洗两遍,并用三氯甲烷萃取,有机相收集后蒸干,得到呈淡黄色粘稠液体的最终产物137.7g,即5,5'
‑ꢀ
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯,硫醇当量为135g/eq,总产率 84.5%。该5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯基本没有硫臭味。
[0100]
该5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯的1h
‑
nmr图以及ir 光谱图分别如图3和图4所示。从图3可以看出,化学位移6.85ppm和7.05ppm附近为联苯环上的质子峰,化学位移4.03ppm、2.59ppm和1.98ppm处的峰对应联苯环上的烷氧基,化学位移2.73ppm、2.49ppm和1.92ppm处的峰则对应联苯环上的烷基,化学位移1.43ppm 和1.31ppm处的峰分别对应联苯环上烷基巯基和烷氧基巯基。从图4可以看出,1489cm
‑1处是联苯环上的吸收峰,818cm
‑1处对应联苯环上的ar
‑
h弯曲振动,2925cm
‑1处为烷基链上的c
‑
h
伸缩振动吸收峰,1242cm
‑1出现的吸收峰为烷氧基c
‑
o伸缩振动,2562cm
‑1处的吸收峰则对应巯基。由此可以看出,该5,5'
‑
双(3
‑
巯基丙基)
‑
2,2'
‑
双(3
‑
巯基丙氧基)
‑
3,3'
‑
二甲氧基联苯具有式(
ⅴ
)所示的结构,其中,r1、r2、r5和r7均为氢原子,r3和r8分别独立地选自甲氧基,r4和r6均为碳原子数为3的亚烷基,m和n均为1。
[0101][0102]
实施例1
‑
22以及对比例1
‑3[0103]
实施例1
‑
8分别按照下表1中的组分及重量份数准备原材料;
[0104]
实施例9
‑
16分别按照下表2中的组分及重量份数准备原材料;
[0105]
实施例17
‑
22分别按照下表3中的组分及重量份数准备原材料;
[0106]
对比例1
‑
3分别按照下表4中的组分及重量份数准备原材料。
[0107]
表1
[0108][0109]
表2
[0110][0111]
表3
[0112][0113]
表4
[0114][0115]
表1
‑
4中,涉及的组分来源如下:
[0116]
双酚a型环氧树脂为dic株式会社的epiclon exa
‑
850crp,环氧当量为 170
‑
175g/eq;
[0117]
1,4
‑
环己烷二甲醇二缩水甘油醚为昭和电工株式会社的cdmdg,环氧当量为135g/eq;
[0118]
季戊四醇四(3
‑
巯基丙酸酯)为sc有机化学株式会社的pemp;硫醇当量为122g/eq;
[0119]
1,3,4,6
‑
四(2
‑
巯基乙基)甘脲的硫醇当量为95g/eq,其结构如式(
ⅵ
)所示:
[0120][0121]
1,1
‑
(二硫代双乙烷二基)
‑
双[3,4,6
‑
三(2
‑
巯基乙基)甘脲]的硫醇当量为127g/eq,具体按照以下方法制备得到:在反应瓶中加入1,3,4,6
‑
四(2
‑
羟基乙基)甘脲3.18g,室温下搅拌,并滴加亚硫酰氯11.75g,滴完后回流2小时;冷却至10℃,添加水10ml、硫脲3.65g,继续回流搅拌反应12小时;然后冷却至25℃,在氮气气氛下滴加48%氢氧化钠水溶液4.00g,70℃搅拌反应9小时;再冷却至20℃,添加浓盐酸3.50g、氯仿10ml,搅拌30分钟,之后进行第1次抽吸过滤,在所得滤饼中添加氯仿10ml,搅拌30分钟,然后进行第2次抽吸过滤。将两次抽吸过滤的滤液合并后除去水层,有机层用5ml水清洗5次,有机层减压浓缩,得到3.1g的黄色油状物(粗产物);利用柱层析(洗脱液:氯仿)对该粗产物进行分离纯化,得到2.85g白色晶体(熔点:75.3℃
‑
77.8℃),即为1,3,4,6
‑
四(2
‑
巯基乙基)甘脲;另外得到0.28g 淡黄色油状物,即为产物1,1'
‑
(二硫代双乙烷二基)
‑
双[3,4,6
‑
三(2
‑
巯基乙基)甘脲]。
[0122]
潜在性固化促进剂为旭化成株式会社的novacure hxa9322hp;
[0123]
硼酸三异丙酯为萨恩化学技术(上海)有限公司的w330012;
[0124]3‑
环氧丙氧基丙基三甲氧基硅烷为信越公司的kbm
‑
403;
[0125]
气相二氧化硅为evonik公司的aerosil r202。
[0126]
实施例1
‑8[0127]
s1:分别按照表1中的组分和重量份数准备原材料;
[0128]
s2:将s1准备好的原材料加入到行星式搅拌机,在室温下初步混合15分钟,然后在室温下使用三辊磨分散30分钟,出料分装,即得到环氧树脂组合物。其中,所述环氧树脂组合物中环氧树脂的环氧官能团当量与硫醇化合物/硫醇系固化剂的硫醇官能团当量之比如下表5所示。
[0129]
实施例9
‑
16
[0130]
s1:分别按照表2中的组分和重量份数准备原材料;
[0131]
s2:将s1准备好的原材料加入到行星式搅拌机,在室温下初步混合15分钟,然后在室温下使用三辊磨分散30分钟,出料分装,即得到环氧树脂组合物。其中,所述环氧树脂组合物中环氧树脂的环氧官能团当量与硫醇化合物/硫醇系固化剂的硫醇官能团当量之比如下表5所示。
[0132]
实施例17
‑
22
[0133]
s1:分别按照表3中的组分和重量份数准备原材料;
[0134]
s2:将s1准备好的原材料加入到行星式搅拌机,在室温下初步混合15分钟,然后在室温下使用三辊磨分散30分钟,出料分装,即得到环氧树脂组合物。其中,所述环氧树脂组合物中环氧树脂的环氧官能团当量与硫醇化合物/硫醇系固化剂的硫醇官能团当量之比如下表5所示。
[0135]
对比例1
‑3[0136]
s1:分别按照表4中的组分和重量份数准备原材料;
[0137]
s2:将s1准备好的原材料加入到行星式搅拌机,在室温下初步混合15分钟,然后在室温下使用三辊磨分散30分钟,出料分装,即得到参比环氧树脂组合物。其中,所述环氧树脂组合物中环氧树脂的环氧官能团当量与硫醇化合物/硫醇系固化剂的硫醇官能团当量之比如下表5所示。
[0138]
表5
[0139]
项目环氧官能团当量与硫醇官能团当量之比实施例11.0实施例21.0实施例31.0实施例41.0实施例51.0实施例61.0实施例71.0实施例81.0实施例91.0
实施例101.0实施例111.0实施例121.0实施例131.0实施例141.0实施例151.0实施例161.0实施例170.6实施例181.5实施例191.8实施例200.7实施例211.5实施例221.8对比例11.0对比例21.0对比例31.0
[0140]
测试例
[0141]
(1)晶体析出时间的测定:
[0142]
分别将实施例1
‑
22与对比例1
‑
3中制备的环氧树脂组合物在室温下放置,从树脂组合物制备完成直至确认晶体析出为止的时间。需要说明的是,晶体析出的确认通过目视进行,测试最长时间为240小时。
[0143]
(2)玻璃化转变温度的测定:
[0144]
使用美国ta仪器的q
‑
800型动态热机械分析测试仪(dma)进行测试,分别将实施例1
‑
22与对比例1
‑
3中制备的环氧树脂组合物,室温下密封静置储存240小时后,再分别取出制备样品,然后置于80℃烘箱中热固化60分钟,将固化完全的树脂组合物制成 42mm
×
8mm
×
0.3mm的薄片,在
‑
40~250℃的温度范围内,在液氮氛围和薄膜拉伸模式下测定损耗因子(tanδ)随温度的变化规律,其中,升温速率10℃/min,测试频率为10hz,从而确定树脂组合物固化后的玻璃化转变温度(℃)。
[0145]
(3)热粘接强度的测定:
[0146]
将实施例1
‑
22与对比例1
‑
3中制备的树脂组合物,室温下密封静置储存240小时后,再分别取出制备样品,将树脂组合物涂覆在喷砂处理后的铝片基材上,用另一个铝片搭接压合,制作试验样品,粘接面积为25.4mm
×
5mm,并保证胶层的厚度为0.1mm,在80℃/60 分钟的条件下进行固化,然后将固化完全的样品,使用万能试验机将两个片材沿相反方向拉开,在环境温度为85℃的条件下进行测试,所测得的力值以强度(mpa)记录;将固化后的样品经过加热加湿条件85℃/85%rh处理120小时后,再次在环境温度为85℃的条件下测试样品的剪切粘接强度(mpa)并记录。
[0147]
上述晶体析出时间、玻璃化转变温度以及加热加湿前后的热粘接强度的测定结果如下表6所示。
[0148]
表6
[0149][0150]
结合表1
‑
6分析比较实施例1
‑
22和/或对比例1
‑
3,首先通过对实施例1
‑
16进行分析可以发现,本发明提供的环氧树脂组合物的晶体析出时间均大于240小时,树脂组合物在静置储存240小时再固化后的玻璃化转变温度均超过了115℃,在85℃高温下测定出的热粘接强度均达到了8.8mpa以上,经过加热加湿试验后的热粘接强度仍然能保持在7.9mpa以上,说明本发明提供的环氧树脂组合物均具有出色的储存稳定性、耐热性、粘接强度和耐湿热性能;对实施例17
‑
22进行分析可以发现,环氧官能团当量与硫醇官能团当量的比值对于
树脂组合物的储存稳定性和耐湿热性能没有明显影响,树脂组合物在240小时以内均没有晶体析出,树脂组合物固化后经过加热加湿实验前后热粘接强度的降低幅度依然比较小,环氧官能团当量与硫醇官能团当量的比值对于树脂组合物的粘接性能有一定的影响,该比值最优为0.9
‑
1.2,当该比值过大或过小时,树脂组合物固化后的热粘接强度有小幅度的下降,但依然达到7.5mpa以上,玻璃化转变温度也有一定的降低,但整体上仍然超过了 100℃。
[0151]
通过分析实施例1、实施例9和对比例1,可以发现将本发明制备的多硫醇化合物更换为含酯键的多硫醇固化剂季戊四醇四(3
‑
巯基丙酸)酯后,树脂组合物固化后的玻璃化转变温度急剧降低,仅仅只有63℃,同时热粘接强度也明显降低至5.44mpa,特别是加热加湿后的热粘接强度几乎消失殆尽,说明本发明的多硫醇化合物对树脂组合物的耐热性、粘接性能和耐湿热水解性能均有明显地影响。
[0152]
通过分析实施例1、实施例9和对比例2,可以发现将本发明制备的多硫醇化合物更换为多硫醇固化剂1,3,4,6
‑
四(2
‑
巯基乙基)甘脲后,树脂组合物的晶体析出时间大幅缩短至7.0 小时,同时固化物的玻璃化转变温度和热粘接强度也出现了一定程度的下降,说明固体的多硫醇固化剂1,3,4,6
‑
四(2
‑
巯基乙基)甘脲在储存一段时间后析出晶体,固化不完全造成了环氧树脂组合物综合性能的降低。
[0153]
通过分析实施例1、实施例9和对比例2
‑
3,可以发现,在对比例3中通过加入部分液态的1,1
‑
(二硫代双乙烷二基)
‑
双[3,4,6
‑
三(2
‑
巯基乙基)甘脲]改善了固体多硫醇固化剂 1,3,4,6
‑
四(2
‑
巯基乙基)甘脲晶体析出时间短的问题,但这种方案在明显增加成本的同时,树脂组合物的玻璃化转变温度和热粘接强度依然低于本发明的树脂组合物,说明本发明的多硫醇化合物可以在低成本且无晶体析出的情况下对树脂组合物的储存稳定性、耐热性、粘接性能和耐湿热水解性能产生明显地影响。
[0154]
综上所述,本发明提供的环氧树脂组合物使用了硫醇化合物作为固化剂,该硫醇化合物在气味低的基础上,无酯键,具有良好的耐湿耐热性能,在室温下为液体,且可直接作为固化剂,用于树脂组合物的合成,该树脂组合物可作为密封剂和粘接剂的成分,相较于专利cn201680014880.5中提出的两种巯基乙基甘脲化合物联用作为固化剂成分,本发明的多硫醇化合物反应工序简单,不用额外进行偶联形成低聚物混合物,也无须与其他多硫醇化合物联用,成本低。此外,本发明提供的多硫醇化合物能够降低单组份低温固化环氧树脂储存稳定性风险,适用期长,应用前景广泛。
[0155]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在不脱离本发明的原理和宗旨的情况下在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。