一种猪流行性腹泻病毒感染性cdna克隆及其构建方法与应用
技术领域
1.本发明属于生物技术领域,具体涉及猪流行性腹泻病毒感染性cdna克隆的构建方法及其应用。
背景技术:
2.猪流行性腹泻(porcine epidemic diarrhea,ped)是由猪流行性腹泻病毒(porcine epidemic diarrhea virus,pedv)引起的一种急性和高度传染性的猪肠道疾病,临床症状与猪传染性胃肠炎病毒(transmissible gastroenteritis virus,tgev)相似,其特征是呕吐、腹泻和厌食,最后因消瘦和严重脱水死亡。各年龄段均可感染,其中仔猪感染后的临床症状尤为严重,死亡率接近100%。除了感染哺乳仔猪,pedv还可降低育肥猪的生长性能。近年来,高致病性pedv变异毒株的出现和广泛传播给养猪业造成了巨大的经济损失。除生物安全防控外,疫苗接种是防控猪流行性腹泻最有效的方法。但是,目前商品化的疫苗提供的保护有限,研制新一代疫苗仍然迫在眉睫。pedv归属于套式病毒目、冠状病毒亚科、甲型冠状病毒属。它是一种囊膜包裹的单股正链rna病毒,其全长基因组约28kb,包括5’端帽子结构和非编码区,3’端非编码区和poly(a)尾巴,以及7个开放阅读框(orfs),包括orf1a、orf1b、s、orf3、e、m和n基因。5’端的orf1a和orf1b编码的两个多聚蛋白(pp1a和pp1ab),3’端的5个orf依次编码纤突蛋白(s)、orf3蛋白、小包膜糖蛋白(e)、膜糖蛋白(m)和核衣壳蛋白(n)。有研究表明,orf3为pedv复制非必需元件,可被替换用于表达外源基因。
3.反向遗传操作系统是一种定向改造和修饰病毒基因组的重要基因工程技术,该技术已成为病毒学基础研究和疫苗研发必不可少的技术平台。目前,pedv反向遗传操作系统主要包括以下三种:基于靶向rna重组的技术、基于细菌人工染色体的感染性cdna克隆、基于体外连接的感染性cdna克隆等。根据病毒拯救方式不同,pedv反向遗传操作系统又可以分为rna转染和dna转染两类系统。为简化pedv感染性cdna克隆的构建过程,本发明以基因ii型的pedv hm毒株为亲本株,基于酵母转化同源重组技术(transformation associated recombination,tar)构建了cmv启动的猪流行性腹泻病毒的反向遗传操作平台,并借助crispr/cas9基因编辑技术构建了表达绿色荧光蛋白的pedv标记病毒。本发明为pedv致病机制及其基因工程疫苗研制奠定了基础。
技术实现要素:
4.发明目的:本发明需要解决的技术问题是提供一种猪流行性腹泻病毒的感染性cdna克隆的构建方法,该构建方法简便、稳定好、效率高,该感染性克隆拯救的病毒rpedv和rpedv-egfp感染vero ccl-81细胞后能够引起细胞病变,生长特性与亲本病毒基本一致,且保持遗传稳定。
5.技术方案:为解决上述技术问题,本发明提供如下技术方案:
6.一种猪流行性腹泻病毒感染性cdna克隆的构建方法,包括如下步骤:
7.(1)以猪流行性腹泻病毒的cdna为模板、seq id no.:1~14所示的序列为引物,将
猪流行性腹泻病毒生物基因组分为7个片段扩增出来;
8.(2)以pyes1l-vector为模板、seq id no.:15~16所示的序列为引物,扩增得到线性化载体pyes1l,所述pyes1l-vector的核苷酸序列如seq id no.:18所示;
9.(3)将线性化载体pyes1l与步骤(1)得到的7个基因组片段混和,加入到mav203酵母感受态中,利用醋酸锂转化法转化至酵母细胞,接种于色氨酸(trp)缺陷型平板上,经菌落pcr法筛选出阳性克隆,将阳性克隆的酵母裂解液转化dh10b电击感受态,获得重组质粒,即猪流行性腹泻病毒感染性cdna克隆。
10.作为优选,所述猪流行性腹泻病毒的基因组序列如seq id no.:17所示。
11.上述猪流行性腹泻病毒感染性cdna克隆的构建方法构建得到的猪流行性腹泻病毒感染性cdna克隆。
12.基于crispr/cas9基因编辑技术的表达绿色荧光蛋白的猪流行性腹泻病毒构建方法,包括以下步骤:
13.(4)以egfp基因为模板,以seq id no.:19~20所示的序列为引物进行pcr扩增,得到egfp基因片段;
14.(5)用cas9 nuclease和两条guide rna切割权利要求1得到的猪流行性腹泻病毒感染性cdna克隆,所述guide rna的序列如seq id no.:21~22所示,得到缺失orf3的猪流行性腹泻病毒感染性cdna克隆骨架;
15.(6)将egfp和缺失orf3的猪流行性腹泻病毒感染性cdna克隆骨架片段混合,进行体外同源重组,转化至dh10b电击感受态中,获得重组质粒,即表达绿色荧光蛋白的猪流行性腹泻病毒。
16.上述基于crispr/cas9基因编辑技术的表达绿色荧光蛋白的猪流行性腹泻病毒构建方法构建得到的表达绿色荧光蛋白的猪流行性腹泻病毒在本发明的保护范围之内。
17.上述猪流行性腹泻病毒感染性cdna克隆或表达绿色荧光蛋白的猪流行性腹泻病毒在拯救病毒中的应用。
18.进一步地,利用转染试剂lipofectamine
tm 3000将权利要3猪流行性腹泻病毒感染性cdna克隆和辅助质粒共转染veroccl-81细胞,当出现典型细胞病变收取病毒上清,获得拯救的rpedv。
19.作为优选,所述辅助质粒的构建方法如下:以猪流行性腹泻病毒感染性cdna克隆为模板、seq id no.17~18为引物,pcr扩增得到n基因片段,n基因片段插入到pcaggs载体的saci和xhoi位点之间,得到辅助质粒。
20.下面以猪流行性腹泻病毒hm株为例对本发明的方法进行进一步说明。
21.猪流行性腹泻病毒hm株感染性cdna克隆构建方法,包括以下步骤:
22.(1)以pyes1l载体为模板,用引物pyes1l-f和pyes1l-r进行pcr扩增,得到线性化的pyes1l载体;
23.(2)以猪流行性腹泻病毒hm株的病毒rna的反转录产物为模板,用引物pedv-f1和pedv-r1扩增f1片段,用引物pedv-f2和pedv-r2扩增f2片段,用引物pedv-f3和r3扩增f3片段,用引物pedv-f4和pedv-r4扩增f4片段,用引物pedv-f5和pedv-r5扩增f5片段,用引物pedv-f6和pedv-r6扩增f6片段,采用引物pedv-f7和pedv-r7扩增f7片段;
24.(3)将线性化载体pyes1l和f1~f7片段混和,导入酵母内进行同源重组,得到猪流
行性腹泻病毒感染性cdna克隆质粒pyes1l-pedv。
25.基于crispr/cas9基因编辑技术的表达绿色荧光蛋白的猪流行性腹泻病毒构建方法,包括以下步骤:
26.(7)以pegfp-n1为模板,用引物pedv-egfp-f和pedv-egfp-r进行pcr扩增,得到egfp基因片段;
27.(8)用cas9 nuclease和两条guide rna(pedv-sgrna 1和pedv-sgrna 2)切割pyes1l-pedv质粒,得到缺失orf3的猪流行性腹泻病毒感染性cdna克隆骨架;
28.(9)将egfp和缺失orf3的猪流行性腹泻病毒感染性cdna克隆骨架片段混合,进行体外同源重组,转化至dh10b电击感受态中,获得重组质粒pyes1l-pedv-egfp。
29.本发明中,所述应用包括如下步骤:构建表达猪流行性腹泻病毒hm毒株n蛋白的辅助质粒,利用转染试剂lipofectamine
tm 3000将感染性cdna克隆质粒(pyes1l-pedv或pyes1l-pedv-egfp)和辅助质粒共转染vero ccl-81细胞,收获细胞培养物,获得拯救的感染性猪流行性腹泻病毒rpedv或rpedv-egfp。
30.本发明中,所述辅助质粒是将猪流行性腹泻病毒n蛋白的编码基因克隆至pcaggs载体,得到pcaggs-pedv-n辅助质粒。
31.有益效果:
32.与现有技术相比,本发明提供了一种快速、高效、稳定的猪流行性腹泻病毒感染性cdna克隆的构建方法,具有如下优势:
33.(1)本发明提供的感染性cdna克隆构建方法具有高效、快速、稳定等特点。本发明将pedv hm株基因组划分为7个片段,基于酵母同源重组的一步法完成克隆构建,解决了传统的酶切连接效率较低的问题,大大提高了效率。
34.(2)本发明提供的感染性cdna克隆具有良好的稳定性。pyes1l载体含有酵母人工染色体和细菌人工染色体元件,在酵母和细菌中复制具有良好的稳定性,解决了冠状病毒部分复制酶基因在大肠杆菌中不能稳定保存的问题;同时在载体上游有巨细胞病毒(cmv)真核启动子序列,在下游有丁型肝炎病毒核酶(hdv ribozyme)序列与牛生长激素多聚腺苷酸信号(bgh)转录终止序列,大大提高了病毒的拯救效率。
35.(3)本发明提供的感染性cdna克隆可直接利用crispr/cas9基因编辑在基因水平上改造猪流行性腹泻病毒基因组,提高了构建重组病毒的效率,为猪流行性腹泻病毒基因组建立了一个快速、有效的技术平台。
36.(4)本发明提供的感染性cdna克隆直接转染哺乳动物细胞拯救重组病毒。本发明中的感染性cdna克隆包含的cmv启动子可以在经dna转染的细胞中启动病毒基因组rna合成,避免了体外rna转录合成过程,简化了病毒拯救操作流程,也解决了体外转录导致的转录本异质性的问题。
附图说明
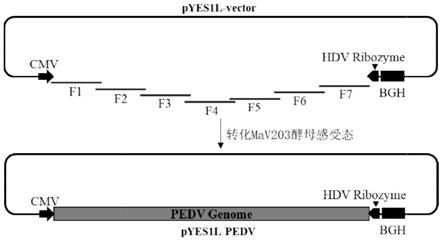
37.图1为猪流行性腹泻病毒感染性cdna克隆的构建示意图。
38.图2为猪流行性腹泻病毒hm株的基因组片段rt-pcr扩增的凝胶电泳图。
39.图3为猪流行性腹泻病毒感染性cdna克隆pyes1l-pedv质粒的限制性内切酶酶切图谱。
40.图4为表达绿色荧光蛋白的猪流行性腹泻病毒感染性cdna克隆pyes1l-pedv-egfp质粒的构建示意图。
41.图5为重组病毒的间接免疫荧光鉴定。
42.图6为感染性cdna克隆拯救的病毒与亲本病毒的多步生长曲线。
43.图7为感染性cdna克隆拯救的病毒与亲本病毒的空斑试验。
具体实施方式
44.下面结合具体实施例对本发明做进一步说明,应该理解的是,这些实施例仅用于例证的目的,决不限制本发明的保护范围。
45.实施例1猪流行性腹泻病毒hm株感染性cdna克隆质粒的构建
46.构建策略如图1所示。根据猪流行性腹泻病毒基因ii型hm毒株全长基因组序列(genbank登录号:mz342899),利用snapgene软件设计7对特异性引物(见表1)分别rt-pcr扩增f1、f2、f3、f4、f5、f6和f7七个片段。
47.按照病毒基因组dna/rna提取试剂盒(天根生化科技有限公司)说明书提取pedv hm株的病毒基因组rna。以病毒基因组rna为模板,按照hm株的病毒基因组rna。以病毒基因组rna为模板,按照iv first-strand cdna synthesis reaction(thermo fisher scientific)说明制备流行性腹泻病毒hm株cdna。反应体系为:2μm gene-specific reverse primer(pedv-r7、pedv-r4或pedv-r2)1μl,10μm dntp mix 1μl,模板rna 11μl。反应程序为:65℃5min,置于冰上1min。之后反应体系为:5
×
ssiv buffer 4μl,100mm dtt 1μl,recombinant rnase inhibitor 1μl,reverse transcriptase(200u/μl)1μl。反应程序为:50℃10min,80℃10min,将至室温。加入1μl rnase h,37℃20min去除rna。
48.扩增f1和f2片段时,以cdna(pedv-r2)为模板,分别用两对引物pedv-f1/pedv-r1和pedv-f2/pedv-r2进行扩增;扩增f3和f4片段时,以cdna(pedv-r4)为模板,分别用两对引物pedv-f3/pedv-r3和pedv-f4/pedv-r4进行扩增;扩增f3和f4片段时,以cdna(pedv-r4)为模板,分别用两对引物pedv-f3/pedv-r3和pedv-f4/pedv-r4进行扩增;扩增f5、f6和f7片段时,以cdna(pedv-r7)为模板,分别用三对引物pedv-f5/pedv-r5、pedv-f6/pedv-r6和pedv-f7/pedv-r7进行扩增;反应体系为:cdna 1μl,上游引物(40μm)和下游引物(40μm)各0.625μl,100mm dntp mix 1μl,5
×
q5 reaction buffer 10μl,q5 high fidelity dna polymerase 0.5μl,ddh2o补至50μl。扩增程序为:98℃30s;98℃10s,58℃-63℃20s,72℃5min,循环30次;72℃2min。目的条带大小分别为4050bp,4122bp,4010bp,4803bp,3417bp,4305bp,3612bp,结果如图2所示。
49.以pyes1l-vector(该质粒由本实验室保存)为模板,用引物pyes1l-f和pyes1l-r进行pcr扩增,获得线性化pyes1l载体。反应体系为:pyes1l-vector 10ng,上游引物(40μm)和下游引物(40μm)各0.625μl,100mm dntp mix 1μl,5
×
q5 rection buffer 10μl,q5 high fidelity dna polymerase 0.5μl,ddh2o补至50μl。扩增程序为:98℃30s;98℃10s,63℃20s,72℃5min,循环30次;72℃2min。目的条带大小应为10243bp。
50.将线性化的pyes1l载体与f1~f7片段混合,通过醋酸锂转化法转化至酵母菌中。具体操作为:各取100ng线性化载体pyes1l与上述片段f1,f2,f3,f4,f5,f6,f7均匀混合;随后从-80℃中取出mav203酵母感受态细胞于30℃中解冻,时间不超过90s,将上述混合物加
入mav203酵母感受态细胞中,轻弹管壁使其混匀,随后加入600μl peg/liac至dna于酵母感受态的混合物中,轻柔上下颠倒混匀后置于30℃水浴中孵育30min,每隔10min轻柔上下颠倒重悬混合物;孵育结束后加入35.5μl的dmso,轻柔上下颠倒混匀后置于42℃水浴中热激20min,每隔5min轻柔上下颠倒混匀;热激结束后1800rpm(200-400xg)离心5min,小心弃去上清,并用1ml 0.9%nacl溶液重悬酵母细胞;取100μl涂于色氨酸缺陷的酵母培养皿中,30℃培养2-3天。由于线性化的pyes1l载体和片段f1、片段f1和片段f2、片段f2和片段f3、片段f3和片段f4、片段f4和片段f5、片段f5和片段f6、片段f6和片段f7、线性化的pyes1l载体和片段f7分别存在着末端同源臂,因此,在宿主细胞酵母内,通过同源重组机制组装成携带cpiv3全长cdna的重组质粒。
51.挑取单菌落置于含15μl lysis buffer的pcr管中,轻轻吹打三次裂解酵母菌,转移5μl至一个新的pcr管中,4℃备用,其余10μl进行菌落pcr检测,筛选出阳性克隆,然后电转化至dh10b电击感受态中。通过spectinomycin抗性筛选阳性重组子,增殖后通过质粒中量提取试剂盒纯化携带pedv全长cdna的重组质粒,命名为pyes1l-pedv。
52.用限制性内切酶mlu i对重组质粒进行酶切鉴定,经0.8%琼脂糖凝胶电泳鉴定酶切图谱。基于snapgene软件预测,酶切图谱应包含4002bp、7175bp,10559bp和16538bp四个片段,结果如图3所示。
53.实施例2基于crispr/cas9基因编辑技术构建重组质粒pyes1l-pedv-egfp
54.构建策略如图4所示。基于crispr/cas9基因编辑技术,设计pedv-sgrna1和pedv-sgrna2(合成于南京金斯瑞生物科技有限公司,见表1)两条guide rna用于切割pyes1l-pedv中orf3的两侧序列,获得缺失orf3的猪流行性腹泻病毒感染性cdna克隆载体骨架。具体操作为:将cas9 nuclease buffer 5μl、pedv-sgrna 1(300nm)和pedv-sgrna 2(300nm)各3μl、cas9 nuclease3μl混合,37℃10min后加入重组质粒pyes1l-pedv 3μg,37℃2h后,回收缺失orf3的猪流行性腹泻病毒感染性cdna克隆载体骨架。
55.以pegfp-n1(clontech)为模板,用引物egfp-f和egfp-r(见表1)进行pcr扩增,得到egfp基因片段。
56.回收纯化后的载体骨架和egfp片段,采用neb公司的hifi dna assembly master mix,通过体外同源重组的方法(参照试剂盒说明书操作)组装重组质粒,电转化dh10b电击感受态细胞,经菌落pcr法筛选出阳性克隆。
57.实施例3:辅助质粒的构建
58.针对pedv中n基因的开放阅读框区域,利用snapgene软件设计特异性引物pedv-n-f和pedv-n-r(见表1),以pyes1l-pedv为模板,进行pcr扩增,得到n基因片段。通过常规酶切连接试验,将回收纯化的n基因片段插入到pcaggs载体的saci和xhoi位点之间,然后将连接产物转化dh5α大肠杆菌感受态细胞,经测序鉴定筛选出正确的重组质粒pcaggs-pedv-n。按照无内毒素质粒提取试剂盒的操作说明提取质粒,测定浓度后分装,于-20℃保存。
59.表1本发明所应用的引物序列
60.[0061][0062]
实施例4:猪流行性腹泻病毒hm株重组病毒及绿色荧光标记病毒的拯救与生物学特性鉴定
[0063]
1.重组病毒的拯救与扩增
[0064]
将vero ccl-81细胞接种于12孔细胞培养板中,待细胞密度达到80%左右,按照lipofectamine 3000转染试剂说明进行质粒转染,每孔的质粒用量为:感染性cdna克隆质粒(重组质粒pyes1l-pedv或pyes1l-pedv-egfp)2μg和辅助质粒pcaggs-n 0.5μg。转染24h后,将细胞培养基换成含有10μg/ml的dmem培养基。
[0065]
待出现典型细胞病变时,收取细胞上清,获得拯救病毒rpedv和rpedv-egfp。将拯救病毒在vero ccl-81细胞连续传代,每一代培养物均置-80℃保存。
[0066]
2.重组病毒的鉴定
[0067]
2.1间接免疫荧光检测病毒n蛋白表达
[0068]
将收取完病毒上清的细胞用4%多聚甲醛固定15min,待细胞完全变干后,加入0.1%triton-x 100覆盖细胞打孔15min,弃去0.1%triton-x 100后加入1%bsa覆盖细胞封闭30min,随后加入抗pedv n蛋白的单克隆抗体,室温孵育1h,用pbs洗5次,再加入alexa488g conjugated oat anti-mouse igg h&l二抗(购自jackson immune research inc),用pbs洗5次,加入dapi染色5min,pbs洗2次,用荧光显微镜观察并记录实验结果(图5)。
[0069]
2.2rt-pcr鉴定
[0070]
分别提取重组病毒(拯救病毒rpedv-egfp)rna,用一步法rt-pcr试剂盒(南京诺唯赞生物科技有限公司),使用引物pedv-egfp-f和pedv-egfp-r进行rt-pcr扩增。
[0071]
1%琼脂糖凝胶电泳后回收片段,送南京金斯瑞生物科技有限公司测序,比较序列是否与设计的一致。结果:扩增的序列与病毒cdna质粒序列一致。
[0072]
3.生物学特性鉴定
[0073]
3.1多步生长曲线
[0074]
将vero ccl-81细胞接种至24孔板中,置于含5%co2的37℃培养箱中培养,待细胞密度达到100%进行感染。将pedv hm、rpedv p5和rpedv-egfp p5以0.01moi分别感染vero ccl-81细胞,每个病毒感染6个孔。分别于感染后0h、24h、48h和72h,收集两个孔的病毒上清,保存于-80℃备用。将收集的病毒上清分别进行10倍梯度稀释,把待感染的vero ccl-81细胞用pbs洗2遍去除培养基中的血清,将稀释好的病毒液加入到96孔板中,每孔加入100μl,各稀释度的病毒设4个重复,孵育2h后弃去培养液,每孔加入100μl含有10μg/ml胰酶的dmem培养基继续在37℃培养。逐日观察并记录病变,于感染后7天观察细胞病变并按照
reed-muench方法计算tcid50,绘制病毒的生长曲线(如图6)。结果表明,与亲本病毒相比,重组病毒(rpedv和rpedv-egfp)在细胞上的生长特性基本一致。但是,在感染后48h和72h rpedv-egfp的病毒滴度比亲本病毒低0.5log。
[0075]
3.2病毒蚀斑试验
[0076]
将vero ccl-81细胞接种至12孔板中,置于含5%co2的37℃培养箱中培养,待细胞密度达到100%进行感染。将pedv hm、rpedv p5和rpedv-egfp p5分别用dmem培养基进行10倍梯度稀释,把待感染的细胞用pbs洗2遍去除培养基中的血清,将稀释好的病毒液加入到12孔板中,37℃、5%co2的条件下孵育。感染2h后,弃去病毒液,将2
×
dmem(含20μg/ml胰酶)和2%ultrapure
tm lmp agarose等比例混匀后加入12孔板中,待完全凝固后倒置于37℃、5%co2培养箱中培养。感染3天后,用4%多聚甲醛固定过夜,第二天弃去4%多聚甲醛与凝胶,每孔加入300μl 1%结晶紫染色液,染色10min,弃去结晶紫溶液、用水清洗,干燥后拍照记录(如图7)。结果表明,三个病毒的空斑形态和大小没有显著差异。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
