1.本发明属于药物领域,具体涉及一种布洛芬衍生物及其制备方法和应用,以及包含所述布洛芬衍生物的药物组合物。
背景技术:
2.布洛芬(ibuprofen),化学名为2-(4-异丁基苯基)丙酸,是一种芳基丙酸类非甾体抗炎药,最早于1968年在英国上市,是一种非选择性环氧化酶抑制剂,它通过抑制环氧化酶,减少前列腺素的合成产生镇痛、抗炎作用;通过下丘脑体温调节中枢而发挥解热作用。因其消炎、镇痛、解热效果好,且不良反应小,故被认为是最安全的非甾体抗炎药(nsaid)之一,目前己被广泛投入临床使用。
3.布洛芬结构中含有一个手性碳,市场上常用的布洛芬为消旋布洛芬,消旋布洛芬中含50%左旋布洛芬和50%右旋布洛芬(即s-布洛芬)。研究表明左旋布洛芬抗炎解热镇痛的作用弱,右旋布洛芬比左旋布洛芬药效强28倍,且左旋布洛芬能引起胃肠道毒性、水钠潴留、肾灌注降低及过敏反应等多种不良反应。
4.目前,上市的右旋布洛芬仅有片剂、胶囊、注射剂和栓剂等,口服制剂的吸收不均匀、首过效应强、生物利用度低,且右旋布洛芬的半衰期短,维持治疗浓度需频繁给药,患者依从性较差,并且会导致发生一些不良反应;目前上市的注射剂静脉给药,注射部位刺激性大。因此,研制新型右旋布洛芬药物,改善并降低其毒副作用,是拓展该药品的临床应用的必然需求。
5.shanbhag等描述了布洛芬等含有羧基的nsaids的胃肠道刺激主要是分子上的羧基引起的,将布洛芬制成前体药物,可提高患者的顺应性[jpharmaceutical sciences,1992,81(2):149-154]。布洛芬前体药物主要集中在对羧基部分进行酰胺化或酯化,由于人体中富含水解酯酶的生物酶,利用布洛芬的羧基进行酯化制备布洛芬衍生物是一种比较有效的方法。
技术实现要素:
[0006]
本发明的目的在于提供一种布洛芬衍生物及制备方法和应用。
[0007]
本发明主要是通过如下技术方案实现的:
[0008]
本发明通过对消旋布洛芬或s-布洛芬的羧基进行衍生化,得到了一种布洛芬衍生物n20及其右旋对映异构体n20(s),克服了布洛芬半衰期短、稳定性差、刺激性和配伍等方面存在的问题。本发明化合物经体外血浆、体内大鼠试验表明具有良好的药代动力学性质。
[0009]
本发明的目的之一是提供一种布洛芬衍生物,即结构式(1)所示的化合物n20,其消旋体、立体异构体、药学上可接受的盐或溶剂合物,或者其药学上可接受的盐的溶剂合物,
[0010][0011]
根据本发明实施方案,其中,结构式(1)所示的化合物选自其右旋对映异构体,其结构如下所示:
[0012][0013]
本发明的目的之二是提供结构式(1)所示的化合物、其消旋体、立体异构体、药学上可接受的盐或溶剂合物或者药学上可接受的盐的溶剂合物的制备方法,包括使化合物1与结构式(2)所示的化合物反应的步骤:
[0014][0015]
其中,在结构式(2)中,x为氯、溴或碘;化合物1为消旋的、s构型或r构型的布洛芬,即为(
±
)-2-(4-异丁基苯基)丙酸、(s)-2-(4-异丁基苯基)丙酸或(r)-2-(4-异丁基苯基)丙酸。
[0016]
根据本发明的实施方案,上述结构式(1)所示的化合物的制备方法在缚酸剂的存在下进行。
[0017]
根据本发明的实施方案,反应温度可以为-5~80℃,反应时间可以为0.5~24h;所用缚酸剂为无机碱如naoh、koh、k2co3、khco3、na2co3、nahco3或有机碱如三乙胺、吡啶、dmap、diea、dbu中的一种、两种或更多种;反应溶剂为丙酮、二氯甲烷、三氯甲烷、四氯化碳、四氢呋喃、甲苯、乙酸乙酯、乙腈、dmf、dmac或乙醚中的一种、两种或更多种。
[0018]
根据本发明的实施方案,所述制备方法具体包括以下步骤:
[0019]
(1)将化合物1溶解在溶剂中,然后加入结构式(2)所示化合物,冰浴下搅拌,分批加入缚酸剂,加毕;
[0020]
(2)将反应容器移至室温搅拌反应过夜,tlc检测反应完全后,向反应容器中加入水、乙酸乙酯、硫代硫酸钠,振摇分液;
[0021]
(3)将反应液萃取,洗涤,有机相干燥后浓缩,经柱层析纯化,得到目标化合物。其中,结构式(2)所示化合物优选为4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮。
[0022]
所述缚酸剂为无机碱如naoh、koh、k2co3、khco3、na2co3、nahco3或有机碱如三乙胺、吡啶、dmap、diea、dbu中的一种、两种或更多种;优选为na2co3、nahco3、三乙胺、吡啶中的一种。
[0023]
所述反应溶剂为丙酮、二氯甲烷、三氯甲烷、四氯化碳、四氢呋喃、甲苯、乙酸乙酯、乙腈、dmf、dmac或乙醚中的一种、两种或更多种;优选为二氯甲烷、甲苯、乙酸乙酯、乙腈、dmf中的一种。
[0024]
本发明的目的之三是提供了上述结构式(1)所示的化合物、其消旋体、立体异构体、药学上可接受的盐或溶剂合物、或其药学上可接受的盐的溶剂合物在制备药物中的应用。
[0025]
根据本发明的实施方案,所述药物可用于治疗一种或更多种以下的疾病:类风湿关节炎、腰痛症、偏头痛、神经痛、肩关节周围炎、骨性关节炎,颈肩腕综合征的消炎和/或镇痛,手术后、外伤后或拔牙后的镇痛和/或消炎、急性上呼吸道炎解热和/或镇痛。
[0026]
根据本发明的实施方案,所述药物为非甾体抗炎药物。
[0027]
本发明的目的四是提供了含有上述结构式(1)所示的化合物、其消旋体、立体异构体、药学上可接受的盐或溶剂合物、或其药学上可接受的盐的溶剂合物的药物组合物。
[0028]
根据本发明的实施方案,所述药物组合物还包含药学上可接受的辅料。所述药物组合物可以为口服制剂,如片剂、胶囊剂、颗粒剂、溶液、混悬液等,还可以为注射剂、滴眼液、凝胶剂、乳膏剂、软膏剂或巴布剂等。
[0029]
有益效果:
[0030]
1、本发明通过对消旋布洛芬的羧基进行衍生化,制备得到了一种布洛芬衍生物,克服了布洛芬半衰期短、稳定性差、刺激性和配伍等方面存在的问题。本发明化合物经体外血浆、体内大鼠试验表明具有良好的药代动力学性质。且化合物本身的理化稳定性较高,在高温试验(60℃放置5~10天)中化合物纯度基本保持不变。
[0031]
2、布洛芬的主要药理活性来源于右旋布洛芬,本发明以右旋布洛芬为原料定向合成了(1s)构型的右旋布洛芬酯前药,研究发现右旋布洛芬酯前药相比左旋布洛芬前药在血浆中的水解速率更快,可生更多的右旋布洛芬。本领域知晓左旋布洛芬抗炎解热镇痛的作用弱,右旋布洛芬(s构型)比左旋布洛芬(r构型)药效强28倍,且左旋布洛芬能引起胃肠道毒性、水钠潴留、肾灌注降低及过敏反应等多种不良反应。因此,研究并制备s构型的布洛芬衍生物即本发明结构式(1)所示的化合物的右旋对映异构体具有重大意义。
附图说明
[0032]
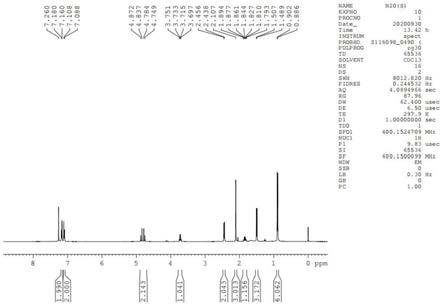
图1为本发明化合物n20(s)的氢谱图谱。
[0033]
图2为本发明化合物n20(s)的质谱图谱。
[0034]
图3为本发明化合物n20(s)在人血浆中的降解速率。
[0035]
图4为本发明化合物n20(s)在人血浆中代谢产生布洛芬的水平。
[0036]
图5为雄大鼠尾静脉给药2mg/kg右布洛芬后测定的右布洛芬平均血药浓度-时间曲线。
[0037]
图6为雄大鼠尾静脉给药3.09mg/kgn20(s)后测定的ibu平均血药浓度-时间曲线。
具体实施方式
[0038]
下面结合具体实施例和附图对本发明作进一步详述,以下实施例只是描述性的,不是限定性的,不能以此限定本发明的保护范围,如无特别说明,所用原料均可通过市售或自制获得。
[0039]
实施例1化合物n20的合成
[0040][0041]
室温下,称取布洛芬(2.1g,10mmol),加入dmf(10ml)搅拌溶解,然后加入4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮(1.5g,10mmol),然后将反应瓶移至冰浴下搅拌,分5批向反应瓶中加入碳酸钠(0.57g,5mmol),加毕。将反应移至室温搅拌反应过夜。tlc检测反应完全后,向反应瓶中加入水100ml,乙酸乙酯50ml,硫代硫酸钠0.5g,振摇分液,有机层在用饱和食盐水洗,无水硫酸钠干燥,抽滤,浓缩,制砂,经快速柱层析得到淡黄色油状物2.0g,收率64%。
[0042]1h nmr(400mhz,cdcl3)δ7.181-7.087(m,4h),4.810(m,2h),3.724(q,j=7,.2hz),2.447(d,j=7.2hz),2.107(s,3h),1.911-1.776(m,1h),1.498(d,j=7.2hz),0.894(d,j=6.6hz).
[0043]
esi-ms m/z=341.1,[m na]
.
[0044]
实施例2:化合物n20(s)的合成
[0045][0046]
实验方法与实施例1相同,区别在于将布洛芬替换为s-布洛芬。
[0047]1h nmr(400mhz,cdcl3)δ7.181-7.087(m,4h),4.810(m,2h),3.724(q,j=7,.2hz),2.447(d,j=7.2hz),2.107(s,3h),1.911-1.776(m,1h),1.498(d,j=7.2hz),0.894(d,j=6.6hz).
[0048]
esi-ms m/z=341.1,[m na]
.
[0049]
实施例3化合物n20(s)的合成
[0050]
室温下,称取s-布洛芬(2.1g,10mmol),加入乙酸乙酯(10ml)搅拌溶解,然后加入4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮(1.5g,10mmol),然后将反应瓶移至冰浴下搅拌,分5批向反应瓶中加入碳酸氢钠(0.57g,5mmol),加毕。将反应移至室温搅拌反应过夜。tlc检测反应完全后,向反应瓶中加入水100ml,乙酸乙酯50ml,硫代硫酸钠0.5g,振摇分液,有机层在用饱和食盐水洗,无水硫酸钠干燥,抽滤,浓缩,制砂,经快速柱层析得到淡黄色油状物2.0g,收率64%。
[0051]
实施例4化合物n20(s)的合成
[0052]
室温下,称取s-布洛芬(2.1g,10mmol),加入dmf(10ml)搅拌溶解,然后加入4-溴甲基-5-甲基-1,3-二氧杂环戊烯-2-酮(1.95g,10mmol),然后将反应瓶移至冰浴下搅拌,分5批向反应瓶中加入三乙胺(0.57g,5mmol),加毕。将反应移至室温搅拌反应过夜。tlc检测反应完全后,向反应瓶中加入水100ml,乙酸乙酯50ml,硫代硫酸钠0.5g,振摇分液,有机层在用饱和食盐水洗,无水硫酸钠干燥,抽滤,浓缩,制砂,经快速柱层析得到淡黄色油状物2.5g,收率80%。
[0053]
实施例5化合物n20(s)的合成
[0054]
室温下,称取s-布洛芬(2.1g,10mmol),加入dmf(10ml)搅拌溶解,然后加入4-碘甲基-5-甲基-1,3-二氧杂环戊烯-2-酮(2.4g,10mmol),然后将反应瓶移至冰浴下搅拌,分5批向反应瓶中加入吡啶(0.57g,5mmol),加毕。将反应移至室温搅拌反应过夜。tlc检测反应完全后,向反应瓶中加入水100ml,乙酸乙酯50ml,硫代硫酸钠0.5g,振摇分液,有机层在用饱和食盐水洗,无水硫酸钠干燥,抽滤,浓缩,制砂,经快速柱层析得到淡黄色油状物2.7g,收率86.4%。
[0055]
试验例1:化合物高温稳定性研究
[0056]
试验方案:将本发明制备的化合物布洛芬衍生物n20适量置于西林瓶中,在高温(60℃)条件下遮光放置,分别于0、5、10天取样,考察化合物纯度及有关物质(布洛芬)变化情况,结果如下表所示:
[0057]
表1纯度测定
[0058][0059]
由试验结果可知,本发明的化合物在高温(60℃)条件下遮光放置10天后,纯度在98%以上说明稳定性良好。
[0060]
试验例2:本发明化合物在人血浆中的代谢研究
[0061]
前体药物是通过在体内酶解释放出的原药发挥疗效,因此,前药在血浆中的代谢
速率与生成速率与其能否有效发挥疗效及延长半衰期密切相关。本发明通过建立体外人体血浆代谢模型对化合物n20及其异构体的转化特性进行评价,实验方案如下:
[0062]
(1)分别配制40mm的化合物n20、n20(s)、n20(r)的纯乙腈贮备液,配制40mm的布洛芬纯乙腈贮备液;
[0063]
(2)取25μl布洛芬贮备液1ml的人血浆中混合,涡旋30s,取样200μl加入800μl乙腈沉降蛋白,涡旋1min终止反应,作为布洛芬对照;40mm的化合物n20、n20(s)、n20(r)贮备液分别稀释200倍作为前药对照;
[0064]
(4)取100μl化合物n20、n20(s)、n20(r)纯乙腈贮备液分别加入4ml的人血浆中混合,涡旋30s,置于37℃恒温振荡水浴加热器中200rpm振荡;
[0065]
(5)在不同时间点(0,15,30,60,120min)取样200μl,每个时间点取样3次,加入800μl乙腈沉降蛋白,涡旋1min终止反应;并同法做空白血浆对照;
[0066]
(6)12000rpm,4℃离心10min,取上清液,(过滤膜)进样30μl,记录峰面积变化;
[0067]
(7)观察并分析化合物n20、n20(s)、n20(r)的水解速率。其中化合物n20(s)经血浆代谢120min后,实验结果如图3和4所示,化合物n20、n20(s)、n20(r)经血浆代谢120min后,实验结果如下表所示:
[0068]
表2化合物n20、n20(s)和n20(r)的代谢
[0069]
化合物前药化合物剩余量(%)活性代谢物生成量(%)n208.72
±
1.5784.52
±
2.68n20(s)0
±
092.67
±
2.12n20(r)14.37
±
1.4980.36
±
2.47
[0070]
由试验结果可知:化合物n20的s型异构体n20(s)代谢速率相比消旋体n20的代谢速率要快,即右旋布洛芬衍生物相比消旋布洛芬衍生物在血浆中的水解速率更快,在体外人血浆中能够比传统消旋布洛芬衍生物更迅速转化为活性代谢物发挥其药理活性作用。试验例3:本发明化合物在大鼠体内药动学研究
[0071]
本发明右布洛芬酯衍生物n20(s)(通过peg400增溶)和右布洛芬(精氨酸盐溶液)对大鼠进行等摩尔剂量尾静脉注射给药后,于给药后5、10、15、30、60、120、240、360、480和720min,眼底静脉取血于肝素处理的试管中。将全血8000rpm离心5min,分取血浆样品置于-80℃下保存,分析测定血浆中右布洛芬的浓度。考察大鼠血浆中右布洛芬与时间的变化关系,经药代动力学软件分析,得药代动力学参数如下表所示:
[0072]
供试品药液配制:大鼠尾静脉给药体积均为0.25ml/100g。
[0073]
受试物右布洛芬:配制含右布洛芬20mg/ml的右布洛芬精氨酸溶液,用移液枪取200μl于10ml ep管中,使用分液枪取4800μl生理盐水加入管中,涡旋混匀,得到浓度为0.8mg/ml,体积为5ml的给药溶液。
[0074]
受试物n20(s):称取右布洛芬酯衍生物n20(s)20mg于15ml ep管中,使用分液枪取10ml的peg 400加入管中,涡旋混匀,得到浓度为2mg/ml的化合物peg溶液。用移液枪取3100μl的n20(s)peg溶液于10ml ep管中,使用分液枪取1900μl生理盐水加入管中,涡旋混匀,配置浓度为1.24mg/ml,体积为5ml的给药溶液。
[0075]
表3给药方案
[0076][0077][0078]
表4药代动力学参数
[0079][0080]
由试验结果可知:右布洛衍生物n20(s)在大鼠体内转化为右布洛芬与右布洛芬盐溶液直接注射后的药时曲线基本重合(见图5和6),auc
0-t
无明显差异,可见该衍生物在大鼠体内能够较快代谢成为具有抗炎活性的右布洛芬,并在大鼠体内整体暴露量一致。药代动力学参数显示,注射类制剂中使用该右布洛芬衍生物代替右布洛芬盐,可以延长右布洛芬的半衰期和平均滞留时间,增大表观分布容积,整体上降低了消除速率。因此,本发明于临床应用中,在达到相同药效的基础上,延长了药物在体内作用时间。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。