1.本发明属于共轭聚合物技术领域,具体来说涉及一种羧酸酯取代聚噻吩衍生物及其制备方法和应用。
背景技术:
2.羧酸酯取代聚噻吩衍生物在光电器件,特别是有机光伏器件中具有广泛的应用,是一类重要的导电高分子材料。
3.目前,这类高分子材料主要通过stille偶联反应制备。该制备方法需要预先合成双官能化的溴化单体和有机锡单体,然后二者等当量反应,得到聚合物。有机锡化合物的合成条件尤为严苛,往往需要极低的温度,无水无氧等。因此,该聚合方法制备羧酸酯取代聚噻吩衍生物存在反应步骤多,耗时,成本高,且在反应过程中会产生化学计量的有毒锡副产物,以及在将其用于光电器件时需要对聚合物进行封端等问题。
4.2019年,b.c.thompson等人(polym.chem.,2019,10,4564-4572)利用直接芳基化法 (direct arylation polymerization,darp),将二羧酸酯功能化的联噻吩与二溴噻吩衍生物反应,制备了一系列羧酸酯取代聚噻吩衍生物,数均分子量可达13.8kda(产率59%)。该方法无需制备有机锡单体,明显缩短了反应步骤,但仍然需要预先制备二溴化单体。
技术实现要素:
5.本发明的目的是针对现有技术中羧酸酯取代聚噻吩衍生物合成方法存在的技术缺陷,而提供一种羧酸酯取代聚噻吩衍生物的制备方法。
6.本发明的另一个目的是提供一种羧酸酯取代聚噻吩衍生物
7.本发明的另一个目的是提供所述羧酸酯取代聚噻吩衍生物的应用。
8.为实现本发明的目的所采用的技术方案是:
9.一种羧酸酯取代聚噻吩衍生物的制备方法,包括以下步骤:
10.在保护气体氛围中,于60~150℃的条件下,单体a、单体b、钯催化剂、氧化剂、添加剂和溶剂反应12-72小时,提纯,得到羧酸酯取代聚噻吩衍生物,
11.所述单体a的结构通式为:
[0012][0013]
其中,r1为烷基或烷基取代基;
[0014]
所述单体b的结构式为:
[0015][0016]
其中,r2为烷基或烷基取代基;
[0017]
r3、r4、r5、r6、r7、r8、r9、r
10
、r
11
为氢、烷基或烷基取代基;
[0018]
x为o、s或n;
[0019]
y为si、c或n;
[0020]
ar为芳烃或杂芳烃。
[0021]
2.如权利要求1所述的羧酸酯取代聚噻吩衍生物的制备方法,其特征在于,所述r1为 c
1-c
30
烷基或c
1-c
30
烷基取代基,r3、r4、r5、r6、r7、r8、r9、r
10
、r
11
、r
12
、r
13
、r
14
和r
15
为氢、c
1-c
30
烷基或c
1-c
30
烷基取代基;
[0022]
所述
[0023][0024]r12
、r
13
、r
14
和r
15
为氢、烷基或烷基取代基,优选为c
1-c
30
烷基或c
1-c
30
烷基取代基;
[0025]c1-c
30
烷基取代基中的取代基可以选自下列基团:环烷基、芳基、杂芳基、杂脂环基、羟基、烷氧基、芳氧基、巯基、烷硫基、芳硫基、卤代、羰基、硫代羰基、o-氨基甲酰基、 n-氨基甲酰基、o-硫代氨基甲酰基、n-硫代氨基甲酰基、c-酰胺基、n-酰胺基、s-亚磺酰氨基、n-亚磺酰氨基、c-羧基、o-羧基、氰硫基、硝基、三卤代甲烷磺酰基或甲硅烷基。
[0026]
在上述技术方案中,所述保护气体氛围为氮气、氩气或其他惰性气体。
[0027]
在上述技术方案中,所述溶剂为甲苯、1,4-二氧六环、邻二甲苯、四氢呋喃、n,n-二甲基乙酰胺、n,n-二乙基甲酰胺、n,n-二乙基乙酰胺、n,n-二甲基甲酰胺、甲苯和二甲基亚砜中的一种或几种的混合物;
[0028]
所述氧化剂为ag2co3、agf、agoac、ag2o、cu(oac)2、cu(otf)2、cucl2和agno3中的一种;
[0029]
所述添加剂为碱、pph3、pcy3hbf4、p(t-bu)3、pivoh(新戊酸)和hoac中的一种;
[0030]
所述碱为na2co3、k2co3、cs2co3、nahco3、naoac、koac、csoac和kf中的一种;
[0031]
所述钯催化剂为pd(dppf)cl2、pd(oac)2、pdcl2、pd2(dba)3、pd(tfa)2、pdcl2(pph3)2、 pd(oh)2、pd(pph3)4和钯碳中的一种。
[0032]
在上述技术方案中,按物质的量份数计,所述单体a、单体b、钯催化剂、氧化剂、碱和添加剂的比为1:1:(0.05~0.2):(1~4):(1~4):(0~4)。
[0033]
在上述技术方案中,所述碱为k2co3、cs2co3或koac,所述氧化剂为ag2co3或 cu(oac)2,所述钯催化剂为pd(oac)2或pd(pph3)4或反式-双[2-(二邻甲苯基膦)苄基乙酸二钯,所述溶剂为四氢呋喃或n,n-二甲基乙酰胺。
[0034]
在上述技术方案中,反应时,于60~150℃条件下,搅拌回流。
[0035]
在上述技术方案中,所述单体a和单体b在所述溶剂中的浓度各为0.01~1mol/l,优选为0.1~0.5mol/l。
[0036]
在上述技术方案中,所述提纯采用索氏提纯,更进一步的,通过以下步骤进行:将所述反应液逐滴加入至甲醇中进行沉降,抽滤,得到粗产品,将所述粗产品先后依次用甲醇和正己烷索氏抽提,再用氯仿索氏抽提,收集氯仿提取液,浓缩后加入甲醇进行沉降,抽滤,干燥后得到羧酸酯取代聚噻吩衍生物。
[0037]
本发明的另一方面,通过以上制备方法得到的羧酸酯取代聚噻吩衍生物。
[0038]
本发明的另一方面,所述羧酸酯取代聚噻吩衍生物作为活性材料在光电器件中的应用。优选的,所述光电器件为光伏器件。
[0039]
与现有技术相比,本发明的有益效果是:
[0040]
1.本发明所需原料简单易得,不需要对原料进行预处理,可以直接进行聚合,避免了使用对空气敏感的试剂(如正丁基锂等)、有毒金属试剂以及产生有毒副产物等;
[0041]
2.该聚合方法可以兼容各种官能团,如酯基、羰基、酰胺基等,适用于多种聚噻吩衍生物的制备;
[0042]
3.制得的聚合物在后续光电器件的使用中,无需进行封端处理,可直接使用,且有良好的光伏性能。
附图说明
[0043]
图1是实施例7所得羧酸酯取代聚噻吩衍生物的光学吸收图谱。
[0044]
图2是实施例7所得羧酸酯取代聚噻吩衍生物的电化学循环伏安曲线图。
具体实施方式
[0045]
以下结合具体实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0046]
反应的一般过程如下所示,将单体a,单体b,钯催化剂,氧化剂,添加剂,溶剂加入反应容器,氮气保护下,保持反应体系60~150℃,反应12-72小时,即可聚合物粗产品。
[0047][0048]
聚合物提纯的一般方法:将聚合物粗产品采用索式提取进行提纯。具体步骤为:将1ml 反应液以每秒钟2滴(每滴的体积为1ml)的速度滴入100ml甲醇中进行沉降,沉降24h,抽滤,得到粗产品。将粗产品分别先后用甲醇和正己烷索氏提取24小时,用于除去杂质和低分子量聚合物,最后用氯仿索氏抽提至索氏提取管中氯仿无色,停止抽提,收集氯仿提取
液,浓缩至2ml,滴入60ml甲醇中进行沉降,沉降24h,抽滤,得到共聚物。
[0049]
下述实施例中所涉及仪器的型号如下:
[0050][0051]
下述实施例中涉及原料如下:
[0052]
[0053]
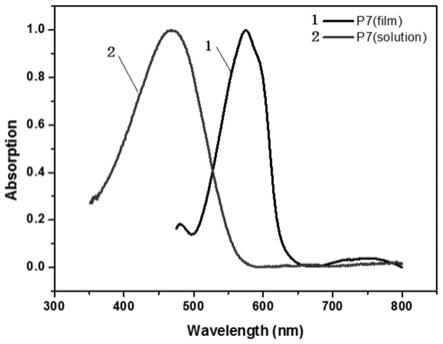
[0054]
[0055][0056]
实施例1:
[0057][0058]
第一步:化合物1的合成
[0059]
向500ml的三颈烧瓶中,加入化合物5-溴噻吩-3-羧酸(38.58mmol),对甲苯磺酸 (77.34mmol)和无水甲醇(200ml),将混合物加热至回流12小时,冷却至室温后,真空除去溶剂,并将残余物溶于二氯甲烷中。将该溶液用水洗涤几次,并用无水硫酸镁干燥,并在真空下除去溶剂。通过用乙醇重结晶来纯化粗产物,得到(5-溴噻吩)-3-羧酸甲酯(7.50g, 88%),为白色晶体。1h nmr(400mhz,cdcl3,δ):7.96(d,j=1.6hz,1h),7.53(d,j=1.2hz, 1h),3.90(s,3h)。
[0060]
第二步:化合物2的合成。
[0061]
将化合物1(20.8mmol),双(频哪醇)二硼酸酯(10.40mmol),三(二亚苄基茚丙酮) 二钯(0)(0.42mmol),三环己基膦氟硼酸盐(1.04mmol),氟化铯(144.00mmol)和无水二恶烷(250ml)加入500ml圆底烧瓶中,将混合物加热至80℃并搅拌24小时。冷却至室温后,将混合物倒入水中并搅拌30分钟,然后用二氯萃取,有机萃取物用盐水洗涤并用无水硫酸镁干燥,并在真空下除去溶剂。粗产物通过硅胶快速柱色谱纯化,用石油醚/二氯甲烷 (1:1)作为洗脱剂,然后用乙醇/三氯甲烷(10:1)重结晶,得到[2,2'-联噻吩]-4,4'-二羧酸二甲酯(2.50g,85%)白色晶体。1h nmr(400mhz,cdcl3,δ):8.00(d,j=1.6hz,2h),7.59(d,j=1.2hz,2h),3.90(s,6h)。
[0062]
第三步:聚合物p1的合成。
[0063]
将[2,2'-联噻吩]-4,4'-二羧酸二甲酯(0.1mmol)、3,3
”-
二辛基-2,2':5',2
”-
三联噻吩 (0.1mmol)、醋酸铜(4eq)、乙酸钾(4eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.1eq) 依次加入到反应器中,取1ml四氢呋喃将上述原料溶解,升温至110℃,在磁力搅
拌条件下反应24h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12 h,即得[2,2'-联噻吩]-4,4'-二羧酸二甲酯-3,3
”-
二辛基-2,2':5',2
”-
三联噻吩共聚物。(34mg,产率43%,mn=4.6kda)。1h nmr(400mhz,cdcl3)δ7.63(d,1h),7.52(s,1h),7.38(s,2h),7.15 (s,1h),7.02(s,1h),3.88(d,6h),2.60(d,4h),1.70(s,4h),1.27(m,20h),0.90
–
0.79(m,6h)。
[0064]
实施例2:
[0065][0066]
化合物3和化合物4合成方法同上述化合物1和化合物2的合成步骤,除了将甲醇换为乙醇。
[0067]
聚合物p2的合成:
[0068]
将化合物4(0.1mmol)、3,3
”-
二辛基-2,2':5',2
”-
三联噻吩(0.1mmol)、碳酸银(2eq)、碳酸钾(2eq)和醋酸钯(0.05eq)依次加入到反应器中,取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至110℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得[2,2'-联噻吩]-4-4'-二羧酸,4,-4'-二乙酯
ꢀ-
3,3
”-
二辛基-2,2':5',2
”-
三联噻吩共聚物。(34mg,产率38%,mn=5.6kda)。1h nmr(400mhz, cdcl3)δ7.63(d,1h),7.52(s,1h),7.38(s,2h),7.15(s,1h),7.02(s,1h),4.20(d,4h),2.80(d,4h), 1.70(s,4h),1.27(m,26h),0.90
–
0.79(m,6h).
[0069]
实施例3:
[0070][0071]
化合物5和化合物6合成方法同上述化合物1和化合物2的合成步骤,除了将甲醇换为 1-正十二醇。
[0072]
聚合物p3的合成:
[0073]
将化合物6(0.1mmol)、2,2'-二噻吩(0.1mmol)、碳酸银(2eq)、碳酸钾(2eq)和反式
ꢀ-
双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)依次加入到反应器中,取1ml四氢呋喃将上述原料溶解,升温至60℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-2,2'
-ꢀ
二噻吩共聚物。(53mg,产率60%,mn=9.7kda)。1h nmr(400mhz,cdcl3)δ7.82(s,1h), 7.49(d,3h),7.07(d,2h),4.29(s,4h),1.74(s,4h),1.26(m,40h),0.86(m,6h).
[0074]
实施例4:
[0075][0076]
第一步:合成化合物7。
[0077]
将2-丁基辛醇(11.59mmol)和5-溴噻吩-3-甲酸(9.66mmol)溶于30ml chcl3中,随后加入4-二甲氨基吡啶(4.83mmol)和n’n-羰基二咪唑(9.66mmol),在80℃下反应10 小时。用氯仿萃取,无水硫酸钠干燥,旋蒸除去溶剂,粗产品柱层析,石油醚作为淋洗液,冷藏后得透亮白色固体(1.6g,44%)。1h nmr(400mhz,cdcl3)δ7.99(d,j=1.3hz,1h),7.57 (d,j=1.3hz,1h),4.19(d,j=5.7hz,2h),1.75(dd,j=11.5,5.7hz,1h),1.33(ddd,j=16.5, 13.8,7.6hz,17h),0.89(dt,j=12.3,6.9hz,6h).
[0078]
第二步:合成化合物8。
[0079]
将化合物7(5.33mmol)和联硼酸频哪醇酯(2.67mmol)溶于30ml二甲基亚砜中,随后加入乙酸钾(15.9mmol)和[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物 (0.16mmol),在80℃下反应8小时。用乙酸乙酯萃取,该反应会有严重的乳化现象,利用饱和氯化钠破乳,无水硫酸钠干燥,旋蒸除去溶剂,粗产品柱层析,石油醚/乙酸乙酯=4:1,冷藏后得透亮白色固体(0.9g,58%)。1h nmr(400mhz,cdcl3)δ7.99(d,j=1.3hz,2h),7.57 (d,j=1.3hz,1h),4.19(d,j=5.7hz,4h),2.03
–
1.62(m,2h),1.33(ddd,j=16.5,13.8,7.6hz, 34h),0.89(dt,j=12.3,6.9hz,13h).
[0080]
第三步:聚合物p4的合成。
[0081]
将化合物8(0.2mmol)、噻吩(0.2mmol)、碳酸银(4eq)、乙酸钾(24eq)和反式-双 [2-(二邻甲苯基膦)苄基乙酸二钯(0.1eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至 90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在 40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-噻吩共聚
物。(40mg,产率40%,mn=18.9kda)。1h nmr(400mhz,cdcl3)δ7.63(s,1h),7.56(s,1h),7.50(s,1h), 4.20(d,2h),4.06(s,4h),1.68(s,2h),1.33
–
1.11(m,35h),0.86(m,12h).
[0082]
实施例5:
[0083][0084]
聚合物p5的合成:
[0085]
化合物8(0.1mmol)、3-辛基噻吩(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和反式
-ꢀ
双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-3-辛基噻吩共聚物。(34mg,产率41%,mn=7.8kda)。1h nmr(400mhz,cdcl3)δ7.62(s,1h),7.53(d,1h), 7.44(s,1h),4.19(d,4h),2.55(s,2h),1.75(s,2h),1.61(s,2h),1.29(m,42h),0.85(m,15h).
[0086]
实施例6:
[0087][0088]
聚合物p6的合成:
[0089]
化合物8(0.1mmol)、呋喃(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-呋喃共聚物。(24mg,产率 35%,mn=6.8kda)1h nmr(400mhz,cdcl3)δ7.63(s,1h),7.56(s,1h),6.30(s,1h),5.90(s, 1h),4.20(d,4h),2.01(s,2h),1.33
–
1.11(m,32h),0.86(m,12h).
[0090]
实施例7:
[0091]
[0092]
聚合物p7的合成:
[0093]
化合物8(0.1mmol)、2,2'-二噻吩(0.1mmol)、碳酸银(4eq)、乙酸钾(4eq)和反式
-ꢀ
双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在60℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-2,2'-二噻吩共聚物。(42mg,产率53%,mn=16.1kda)。1h nmr(400mhz,cdcl3)δ7.52(s,2h),7.46(s,2h), 7.20(s,2h),4.21(d,4h),1.75(s,2h),1.42
–
1.25(m,33h),0.93
–
0.79(m,13h).
[0094]
实施例8:
[0095][0096]
聚合物p8的合成:
[0097]
化合物8(0.1mmol)、3,3'-二辛基-2,2'-联噻吩(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq) 和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-3,3'-二辛基-2,2'-联噻吩共聚物。(88mg,产率89%,mn=12.4kda)。1h nmr(400mhz,cdcl3)δ7.62 (s,1h),7.53(d,2h),7.44(s,1h),4.19(d,4h),2.58(s,4h),1.75(s,2h),1.61(s,4h),1.29(m, 56h),0.85(m,18h).
[0098]
实施例9:
[0099][0100]
聚合物p9的合成:
[0101]
化合物8(0.1mmol)、3,3
”-
二辛基-2,2':5',2
”-
三联噻吩(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和醋酸钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-3,3
”-
二辛基-2,2':5',2
”-
三联噻吩共聚物。(40mg,产率40%,mn=17.5kda)。1h nmr(400mhz,cdcl3)δ7.63(d,1h),7.52(s,1h), 7.38(s,2h),7.15(s,1h),7.02(s,1h),4.20(d,4h),2.80(d,5h),1.70(s,4h),1.27(m,56h),0.90
–ꢀ
0.79(m,18h).
[0102]
实施例10:
[0103][0104]
聚合物p10的合成:
[0105]
化合物8(0.1mmol)、2,5-双(2-辛基十二烷基)-3,6-双(噻吩-2-基)吡咯并[3,4-c]吡咯
ꢀ-
1,4(2h,5h)-二酮(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和反式-双[2-(二邻甲苯基膦) 苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应72h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-2,5-双(2-辛基十二烷基)-3,6-双(噻吩-2
-ꢀ
基)吡咯并[3,4-c]吡咯-1,4(2h,5h)-二酮共聚物。(34mg,产率23%,mn=7.5kda)。1h nmr(400 mhz,cdcl3)δ7.63(d,1h),7.52(s,1h),7.38(s,2h),7.15(s,1h),7.02(s,1h),4.20(d,4h),3.05 (d,4h),1.70(s,4h),1.27(m,96h),0.90
–
0.79(m,24h).
[0106]
实施例11:
[0107][0108]
聚合物p11的合成:
[0109]
化合物8(0.1mmol)、4,4-二(2-乙基己基)-二噻吩并噻咯(0.1mmol)、碳酸银(4eq)、乙酸钾(4eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.1eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-4,4-二(2-乙基己基)-二噻吩并噻咯共聚物。(45mg,产率43%,mn=6.5kda)。1h nmr (400mhz,cdcl3)δ7.68
–
7.38(m,3h),7.05
–
6.88(m,1h),4.21(d,4h),1.93(d,2h),1.25
–ꢀ
1.33(m,56h),0.92
–
0.54(m,24h).
[0110]
实施例12:
[0111][0112]
聚合物p12的合成:
[0113]
化合物8(0.1mmol)、4-(己基癸基)-4h-二噻吩并[3,2-b:2',3'-d]吡咯(0.1mmol)、碳酸银 (4eq)、乙酸钾(4eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.1eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]
ꢀ-
4,4'-二羧酸酯-4-(己基癸基)-4h-二噻吩并[3,2-b:2',3'-d]吡咯共聚物。(30mg,产率29%,mn=3.5 kda)。1h nmr(400mhz,cdcl3)δ7.62(s,1h),7.53(d,2h),7.44(s,1h),4.19(d,4h),3.93 (d,2h),1.75(s,2h),1.61(s,4h),1.29(m,56h),0.85(m,18h).
[0114]
实施例13:
[0115][0116]
聚合物p13的合成:
[0117]
化合物8(0.1mmol)、4,4-二(2-乙基己基)-二噻吩并环戊二烯(0.1mmol)、碳酸银(2eq)、碳酸钾(2eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温 20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-4,4-二(2-乙基己基)-二噻吩并环戊二烯共聚物。(34mg,产率33%,mn=3.6kda)。1h nmr(400mhz,cdcl3)δ7.68
–
7.38(m,3h),7.05
–
6.88(m,1h),4.21(d,4h),1.93(d,2h), 1.84
–
1.53(m,4h),1.19(m,45h),0.92
–
0.54(m,24h).
[0118]
实施例14:
[0119][0120]
聚合物p14的合成:
[0121]
化合物8(0.1mmol)、并三噻吩(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和反式
-ꢀ
双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在40℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-并三噻吩共聚物。 (33mg,产率39%,mn=1.8kda)。1h nmr(400mhz,cdcl3)δ7.69(s,1h),7.47(d,1h),7.35
ꢀ–
7.31(m,1h),7.23(d,1h),4.14(d,4h),1.49
–
1.47(m,5h),1.20(m,33h),0.79(m,12h).
[0122]
实施例15:
[0123][0124]
聚合物p15的合成:
[0125]
化合物8(0.1mmol)、4,8-双[(2-乙基己基)氧基]苯并[1,2-b:4,5-b']二噻吩(0.1mmol)、碳酸银(2eq)、乙酸钾(2eq)和反式-双[2-(二邻甲苯基膦)苄基乙酸二钯(0.05eq)取1ml n,n-二甲基乙酰胺将上述原料溶解,升温至90℃,在磁力搅拌条件下反应48h后,停止反应,冷却至室温20~25℃,得到含有羧酸酯取代的聚噻吩衍生物的反应液。采用索式提取方法对所得反应液进行提取,将提取所得固体在60℃真空干燥箱中干燥12h,即得双甲基-[2,2'-联噻吩]-4,4'-二羧酸酯-4,8-双[(2-乙基己基)氧基]苯并[1,2-b:4,5-b']二噻吩共聚物。(40mg,产率38%,mn=7.7kda)。1h nmr(400mhz,cdcl3)δ7.90(d,1h),7.64(t,3h),7.36(d,1h),4.21 (d,4h),4.03(d,3h),1.85(s,2h),1.15(m,46h),1.00
–
0.73(m,24h).
[0126]
实施例16:
[0127]
本发明的另一目的是提供上述制备方法获得的羧酸酯取代聚噻吩衍生物的光伏性能表征。
[0128]
本发明所制备的聚合物可适用于光电器件中的活性材料:如太阳能电池、发光二极管、晶体管等等。且本发明制备的聚合物所用的合成方法是一种新型的方法,具有简洁、
高效、环境友好等特点,有利于降低生产成本和工艺难度。且在太阳能电池上还有一定的应用前景。
[0129]
如上述实施例7中所合成聚合物p7,将p7与各种有机溶剂混合,该有机溶剂包括氯化溶剂,如氯仿,氯苯,二氯苯等等,以及其他溶剂包括甲醇,甲苯,四氢呋喃等等。发现此聚合物在氯化溶剂中具有很好的溶解度,通过将聚合物的氯仿溶液旋涂至玻璃片上二制得一层薄膜。并测其光谱等性质,聚合物的光学带隙经验公式为(e
gopt
=1240/λ,其中eg为聚合物的光学带隙,λ为吸收光谱在长波长方向的起始值)图一为光学吸收数据。
[0130]
聚合物p7在溶液中的最大吸收值位置为462nm,当聚合物旋涂成薄膜后最大吸收值为548nm,从聚合物起始吸收的位置,根据公式egopt=1240/λ,得到聚合物p7光学带隙为 1.85ev,该光学带隙可以提高此器件的光电转换效率。
[0131]
实施例17:
[0132]
利用电化学循环伏安法的最高占用分子轨道(homo)测量。将在实施例7中所合成聚合物溶解于氯仿中,然后滴加该溶液至工作电极如铂片上,使用0.1mol/l bu4npf6的无水乙腈作为电解质溶液;以玻碳电极作为工作电极,饱和甘汞电极作为参比电极,玻丝为辅助电极的三电极系统。二茂铁/二茂铁盐为内标来测量聚合物的电化学性质。根据公式一计算聚合物的homo和lumo能级:
[0133]
homo=-e(e
onsetox
4.8)(ev)
[0134]
lumo=-e(e
onsetred
4.8)(ev)
[0135]
其中e
ox
和e
red
分别为氧化和还原电位,通过上面的公式计算得出此聚合物的所对应的 homo能级为-5.35ev,对应的lumo能级为-2.92ev。说明聚合物p7侧链加入酯基可以降低homo能级,提高器件的开路电压。
[0136]
实施例18:
[0137]
器件架构为ito/pedot:pss/供体:pc71bm/etl-1/al。将样品分别放在去离子水,丙酮和异丙醇中超声,并将样品分别进行15分钟的超声波处理,清洗涂有氧化铟锡(ito) 的玻璃基板,然后通过吹氮气进行干燥。将pedot:pss薄层以3000rpm的速度旋涂到ito 表面。在150℃下烘烤20分钟后,将基材转移到充满氩气的手套箱中。随后,使用氯仿或邻二氯苯溶液将实施例7中聚合物和pc71bm混合物旋涂一层活性层,然后在不同温度下以可变浓度退火10分钟。etl-1薄层和80nm al层在高真空(《2
×
10-4
pa)下沉积在有源层上。
[0138]
基于实施例7的聚合物和实施例18的操作方法制造并表征聚合物太阳能电池器件,本发明进行了一系列溶剂添加剂的筛选,分别选用dio、1,8-辛二硫醇、苯基萘还有热退火(ta) 等用于提高这些聚合物的光伏性能。结果表明未处理过的设备具有最优的光电转化效率。结果如下表:
[0139][0140]
如表所示。基于p7:pc71bm的器件的最佳条件为薄膜状态下开路电压(voc)值为0.83v,短路电流密度(jsc)值为-6.48ma cm-2
,填充因子(ff)为0.51,光电转化效率(pce)为2.72%。
[0141]
以上所述仅是本发明的优选实施方式,应当指出的是,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。