β-丙氨酰-l-组氨的制备方法
技术领域
1.本发明涉及多肽合成技术领域,尤其涉及β-丙氨酰-l-组氨的制备方法。
背景技术:
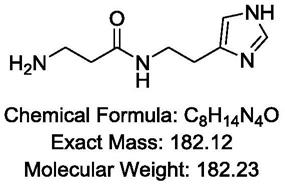
2.脱羧肌肽,化学名为β-丙氨酰-l-组氨(β-alanyl histamine),白色粉末,分子式为c8h
14
n4o
·
2hcl,化学结构式为:
[0003][0004]
β-丙氨酰-l-组氨是一种由β-丙氨酸和l-组氨构成的咪唑类二肽。1975年,β-丙氨酰-l-组氨最早在甲壳类动物体内被发现,随后在一些哺乳动物心脏中也有发现。该成分与肌肽结构类似,具有抗氧化功能,抗糖化功能,因为不会被体内的酶所识别而损失活性,所以具有更好的稳定性。1975年,β-丙氨酰-l-组氨最早在甲壳类动物体内被发现。
[0005]
1994年,exsymol monaco首次将β-丙氨酰-l-组氨应用于化妆品领域,作为化妆品功能添加剂,在分子生物学、细胞、皮肤及临床进行了多方位研究。exsymol相关发表文献显示,β-丙氨酰-l-组氨在皮肤中的抗氧化、氧化逆转、抗污染、抗老化。皮肤紧致,提供细胞能量、抗糖化及糖化逆转等方面显示出优秀的功能特性,特别是去糖化、抗糖毒、逆转糖化反应中间体糖基乙胺作用明显。
[0006]
2008年,学者用分子模型法考察了β-丙氨酰-l-组氨对比肌肽的透皮效果及抗水解能力,β-丙氨酰-l-组氨显示出更好的透皮能力和稳定性。
[0007]
有研究指出,β-丙氨酰-l-组氨的咪唑基含有柔性乙烯基侧链,对于组胺h3受体-配体有重要的相互作用。从一些结果来看,脑组胺与β-丙氨酰-l-组氨之间存在一定的关系,在神经药物学中,β-丙氨酰-l-组氨将是一种新的组胺h3受体拮抗剂被研究。
[0008]
目前报道的合成β-丙氨酰-l-组氨的方法,有些合成步骤长,后处理不方便;有些则需要使用到缩合剂,反应中涉及的β-丙氨酸需要把氨基保护,缩合完成后再脱去,总体路线长,导致总收率不高,成本较高。可见,目前尚缺少适宜大规模生产β-丙氨酰-l-组氨的方法。
技术实现要素:
[0009]
有鉴于此,本发明要解决的技术问题在于提供β-丙氨酰-l-组氨的制备方法,一起提高β-丙氨酰-l-组氨的收率和纯度。
[0010]
本发明提供的β-丙氨酰-l-组氨的制备方法,包括:
[0011]
步骤1:将β-丙氨酸溶液与固体三光气溶液反应,制得1,3-恶嗪烷-2,6-二酮;
[0012]
步骤:2:将组胺二盐酸盐和碳酸钠的溶液与1,3-恶嗪烷-2,6-二酮溶液反应,获得
l-组氨。
[0034]
一些实施例中,所述甲醇水溶液中,甲醇的体积分数为83.33%。
[0035]
一些实施例中,所述酸性环境的ph值为3。一些具体实施例中,由稀盐酸调节ph值。
[0036]
本发明提供的β-丙氨酰-l-组氨的制备方法包括:将β-丙氨酸与固体三光气反应制备1,3-恶嗪烷-2,6-二酮后再开环制得β-丙氨酰-l-组氨。该方法路线缩短至两个步骤,原料转化率高,后处理简单,且得到产品的纯度高。
具体实施方式
[0037]
本发明提供了β-丙氨酰-l-组氨的制备方法,本领域技术人员可以借鉴本文内容,适当改进工艺参数实现。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明。本发明的方法及应用已经通过较佳实施例进行了描述,相关人员明显能在不脱离本发明内容、精神和范围内对本文的方法和应用进行改动或适当变更与组合,来实现和应用本发明技术。
[0038]
本发明提供的β-丙氨酰-l-组氨的制备方法包括制备1,3-恶嗪烷-2,6-二酮和制备β-丙氨酰-l-组氨两个步骤。具体为:
[0039]
1,3-恶嗪烷-2,6-二酮的制备(i)
[0040]
在反应器中加入β-丙氨酸1倍和四氢呋喃5-10倍(重量比),搅拌均匀后在室温下滴加入固体三光气与四氢呋喃的混合液,固体三光气的加入量为1.15-1.35当量,溶解固体三光气的无水四氢呋喃用量为β-丙氨酸重量5-10倍,滴加完毕后,继续室温下搅拌反应8-14小时,反应结束后,减压除去溶剂四氢呋喃,残余物用乙酸乙酯溶解,乙酸乙酯的用量为β-丙氨酸的重量的2倍量,乙酸乙酯有机相用10%的碳酸氢钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,减压蒸馏除去乙酸乙酯得到残余物1,3-恶秦烷-2,6-二酮粗品,粗品无需纯化,可直接用于下一步反应。
[0041]
β-丙氨酰-l-组氨的制备(ii)
[0042]
在反应器中加入组胺二盐酸盐1.1-1.3当量,碳酸钠1.5-2.8当量,水5-10倍(重量比),搅拌至溶清后滴加1,3-恶嗪烷-2,6-二酮1当量与乙腈5-10倍(重量比)混合液,搅拌均匀后升温至30℃反应24-36小时,无二氧化碳溢出,反应结束后减压蒸馏除去溶剂乙腈和水,将残余物倒入2倍量乙酸乙酯中,打浆过滤得到固体,为β-丙氨酰-l-组氨粗品,用稀盐酸调ph=3,经甲醇水结晶得到精品β-丙氨酰-l-组氨。
[0043]
本发明采用的试材皆为普通市售品,皆可于市场购得。下面结合实施例,进一步阐述本发明:
[0044]
实施例1.
[0045]
1,3-恶嗪烷-2,6-二酮的制备
[0046]
在3000ml反应瓶中,加入β-丙氨酸89.1g(1mol)与890ml四氢呋喃,室温下搅拌均匀后滴加入890ml四氢呋喃与固体三光气400.6g(1.35mol)的混合液。滴加完毕后,继续室温下搅拌8小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为180ml,乙酸乙酯有机相用20%的碳酸钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为60.1g,收率为52.28%。
[0047]
实施例2.
[0048]
1,3-恶嗪烷-2,6-二酮的制备
[0049]
在3000ml反应瓶中,加入β-丙氨酸89.1g(1mol)与890ml四氢呋喃,室温下搅拌均匀后滴加入890ml四氢呋喃与固体三光气400.6g(1.35mol)的混合液。滴加完毕后,继续室温下搅拌8小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为180ml,乙酸乙酯有机相用10%的碳酸钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为69.6g,收率为60.47%。
[0050]
实施例3.
[0051]
1,3-恶嗪烷-2,6-二酮的制备
[0052]
在3000ml反应瓶中,加入β-丙氨酸89.1g(1mol)与890ml四氢呋喃,室温下搅拌均匀后滴加入890ml四氢呋喃与固体三光气400.6g(1.35mol)的混合液。滴加完毕后,继续室温下搅拌8小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为180ml,乙酸乙酯有机相用10%的碳酸氢钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为92.5g,收率为80.37%。
[0053]
实施例4.
[0054]
1,3-恶嗪烷-2,6-二酮的制备
[0055]
在2000ml反应瓶中,加入β-丙氨酸50.0g(0.56mol)与500ml四氢呋喃,室温下搅拌均匀后滴加入500ml四氢呋喃与固体三光气191.5g(0.64mol)的混合液。滴加完毕后,继续室温下搅拌10小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为100ml,乙酸乙酯有机相用10%的碳酸氢钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,有机相减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为56.7g,收率为88.1%。
[0056]
实施例5.
[0057]
1,3-恶嗪烷-2,6-二酮的制备
[0058]
在3000ml反应瓶中,加入β-丙氨酸89.1g(1mol)与890ml四氢呋喃,室温下搅拌均匀后滴加入890ml四氢呋喃与固体三光气400.6g(1.35mol)的混合液。滴加完毕后,继续室温下搅拌10小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为180ml,乙酸乙酯有机相用10%的碳酸氢钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为103.6g,收率为90.02%。
[0059]
实施例6.
[0060]
1,3-恶嗪烷-2,6-二酮的制备
[0061]
在2000ml反应瓶中,加入β-丙氨酸50.0g(0.56mol)与500ml四氢呋喃,室温下搅拌均匀后滴加入500ml四氢呋喃与固体三光气191.5g(0.64mol)的混合液。滴加完毕后,继续室温下搅拌14小时,减压除去四氢呋喃。残余物用乙酸乙酯溶解,乙酸乙酯的用量为100ml,乙酸乙酯有机相用10%的碳酸氢钠水溶液洗,水洗,无水硫酸钠干燥后,过滤除去干燥剂,有机相减压蒸馏得到残余物1,3-恶嗪烷-2,6-二酮粗品,粗品为61.2g,收率为95.2%。
[0062]
实施例7.
[0063]
β-丙氨酰-l-组氨的制备
[0064]
在2000ml反应器中加入组胺二盐酸盐202.47g(1.1mol),碳酸钠159.0g(1.5mol),水500ml,搅拌至溶清后滴加1,3-恶嗪烷-2,6-二酮115.0g(1mol,实施例4制得)与乙腈500ml混合液,搅拌均匀后升温至30℃反应24小时,无二氧化碳溢出,反应结束后减压蒸馏
除去溶剂乙腈和水,将残余物倒入240ml乙酸乙酯中,打浆过滤得到固体,为β-丙氨酰-l-组氨粗品,用稀盐酸调至ph=3,经甲醇水结晶得到精品β-丙氨酰-l-组氨191.3g,收率为75.0%。
[0065]
1h nmr(400mhz,d2o)δ8.63(s,1h),7.32(s,1h),3.60-3.56(m,2h),3.30-3.26(m,2h),3.03-2.99(m,2h),2.74-2.68(m,2h).
[0066]
实施例8.
[0067]
β-丙氨酰-l-组氨的制备
[0068]
在2000ml反应器中加入组胺二盐酸盐202.47g(1.1mol),碳酸钠212.0g(2.0mol),水500ml,搅拌至溶清后滴加1,3-恶嗪烷-2,6-二酮115.0g(1mol,实施例3制得)与乙腈500ml混合液,搅拌均匀后升温至30℃反应30小时,无二氧化碳溢出,反应结束后减压蒸馏除去溶剂乙腈和水,将残余物倒入240ml乙酸乙酯中,打浆过滤得到固体,为β-丙氨酰-l-组氨粗品,用稀盐酸调至ph=3,经甲醇水结晶得到精品β-丙氨酰-l-组氨198.7g,收率为77.9%。
[0069]
实施例9.
[0070]
β-丙氨酰-l-组氨的制备
[0071]
在2000ml反应器中加入组胺二盐酸盐202.47g(1.1mol),碳酸钠264.9g(2.5mol),水500ml,搅拌至溶清后滴加1,3-恶嗪烷-2,6-二酮115.0g(1mol,实施例4制得)与乙腈500ml混合液,搅拌均匀后升温至30℃反应36小时,无二氧化碳溢出,反应结束后减压蒸馏除去溶剂乙腈和水,将残余物倒入240ml乙酸乙酯中,打浆过滤得到固体,为β-丙氨酰-l-组氨粗品,用稀盐酸调至ph=3,经甲醇水结晶得到精品β-丙氨酰-l-组氨208.2g,收率为81.6%。
[0072]
实施例10.
[0073]
β-丙氨酰-l-组氨的制备
[0074]
在2000ml反应器中加入组胺二盐酸盐202.47g(1.1mol),碳酸钠296.7g(2.8mol),水500ml,搅拌至溶清后滴加1,3-恶嗪烷-2,6-二酮115.0g(1mol,实施例4制得)与乙腈500ml混合液,搅拌均匀后升温至30℃反应24-36小时,无二氧化碳溢出,反应结束后减压蒸馏除去溶剂乙腈和水,将残余物倒入240ml乙酸乙酯中,打浆过滤得到固体,为β-丙氨酰-l-组氨粗品,用稀盐酸调至ph=3,经甲醇水结晶得到精品β-丙氨酰-l-组氨217.6g,收率为85.3%。
[0075]
以上仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。