1.本发明属于制药领域,主要涉及一种高分散氯氮平原料药晶体颗粒的制备方法。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
3.氯氮平,又称8-氯-11-(4-甲基-1-哌嗪基)-5h-二苯并[b,e][1,4]二氮杂,分子式为c
18h19
cln4,常温下为淡黄色结晶性粉末,无臭,无味,在三氯甲烷中易溶,在乙醇中溶解,在水中几乎不溶,对精神分裂症的阳性或阴性症状产生较好的疗效。
[0004]
目前,氯氮平的制备主要采用冷却结晶的方式,即将其粗品在高温下溶解,然后通过降温来使得药物析出,进而达到提纯的目的。由于氯氮平在水以及一般的有机溶剂中溶解度并不高,所以在采用传统的纯化工艺时,起始的结晶温度往往高于80℃,过高的结晶温度会加速药物主要成分的降解。实践证明,即使采用较大的温度范围来结晶氯氮平药物,其质量收率仍然难以超过50%。另一方面,普通的冷却结晶无法良好控制氯氮平药物晶体颗粒的粒度大小和粒度分布范围,产物存在粒度均一性差、粒度批次差异大、颗粒团聚现象严重等问题,给后期的制剂环节带来了不良影响。
技术实现要素:
[0005]
针对上述所存在的问题,本发明提出了一种高分散氯氮平原料药晶体颗粒的制备方法。通过溶析结晶的方式来制备氯氮平一水合物晶体,制备过程不涉及高温环节。制备过程中通过控制结晶方法来调控晶体的形貌、粒度和粒度分布,通过对氯氮平一水合物干燥环节的调控来控制原料药的晶型。进而得到粒度可控、颗粒分散度高、收率高、晶型纯度高、药学质量指大生产的氯氮平原料药晶体颗粒制备工艺方法。
[0006]
为实现上述技术目的,本发明采用如下技术方案:
[0007]
本发明的第一个方面,提供了一种高分散氯氮平原料药晶体颗粒的制备方法,包括:
[0008]
将氯氮平粗品溶解于有机溶剂中,在20℃下制备其接近饱和的溶液,溶解完全后过滤,滤液收集,在20℃下保温,记为氯氮平溶液;
[0009]
将添加剂溶解在水中,过滤,收集滤液,记为添加剂溶液;
[0010]
向所述氯氮平溶液中滴加添加剂溶液,养晶,得到产物;
[0011]
将所述产物进行过滤、洗涤、干燥,得到氯氮平一水合物晶体;
[0012]
对所述氯氮平一水合物晶体进行干燥处理,得到氯氮平无水物晶体颗粒;
[0013]
其中,所述添加剂为天然高分子化合物。
[0014]
本发明涉及一种高分散度氯氮平原料药晶体颗粒的制备方法,其核心创意在于该方法不涉及高温环节,即通过常温或低温条件下的溶析结晶来制备氯氮平晶体。该方法通
过使用添加剂来调控晶体形貌与粒度,采用干燥方法(先氮吹,然后真空干燥,以含水量来控制干燥终点)来调控氯氮平的晶型,得到的产物具有粒度可控、颗粒分散度高、收率高、晶型纯度高、药学质量指标合格、放大生产容易等优势。
[0015]
本发明的第二个方面,提供了上述的方法制备的氯氮平原料药晶体颗粒,所述氯氮平原料药晶体颗粒的粒度在30-150微米。
[0016]
本发明的第三个方面,提供了天然高分子化合物在制备调控晶体形貌与粒度中的应用。
[0017]
本发明的有益效果在于:
[0018]
(1)本发明的方法可以将氯氮平的收率由不足50%提升到80%以上。显著改善了产物颗粒的团聚问题,同时实现了产品粒度的可控调节。有利于提高氯氮平原料药的可压性。
[0019]
(2)该方法得到的氯氮平原料药晶体颗粒的粒度在30-150微米范围内可控,颗粒分散度高、流动性好、晶型纯度高。该方法操作简单、易于放大生产。
附图说明
[0020]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0021]
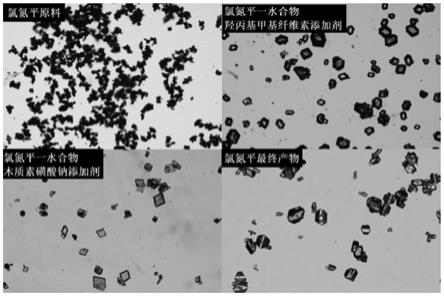
图1:本发明实施例1制备的氯氮平显微镜图片;
[0022]
图2:本发明实施例1制备的氯氮平xrd图谱;
[0023]
图3:本发明实施例1制备的氯氮平粒度分布图;
[0024]
图4:本发明实施例1制备的氯氮平tg图;
[0025]
图5:本发明实施例1制备的氯氮平sem图。
具体实施方式
[0026]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0027]
高分散度氯氮平原料药晶体颗粒的制备方法主要包括以下两个步骤:
[0028]
步骤一:氯氮平一水合物晶体的制备。将10g氯氮平粗品溶解于有机溶剂中,在20℃下制备其接近饱和的溶液,溶解完全后过0.45微米有机滤膜,滤液收集并加入到夹套结晶器中,在20℃下保温。在100g超纯水中加入100ppm的羟丙基甲基纤维素添加剂,使其完全溶解,过0.45微米水性滤膜,滤液待用。开启机械搅拌,用蠕动泵向上述滤液中滴加超纯水,以1ml/min的速度,向结晶器中滴加带有添加剂的超纯水。滴加结束后,恒温搅拌养晶1小时。产物经过滤、去离子水洗涤、真空干燥后得到氯氮平一水合物晶体。通过调节添加剂的种类和含量,可以控制氯氮平一水合物晶体的形貌和平均粒度。
[0029]
此步骤涉及的有机溶剂主要为乙醇、丙酮、n,n-二甲基甲酰胺等。此处提到的添加剂一般为天然高分子化合物,如乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、木质素磺酸钠等。该步骤优选的结晶温度范围为10-25℃,添加剂的添加量为50-1000ppm。本步骤得到的氯氮平一水合物颗粒为立方体或长方体,平均粒度在20-200微米的范围内可控,颗粒
分散度高、收率可达到80%以上。
[0030]
步骤二:氯氮平无水物原料药晶体的制备。即通过对步骤一得到的氯氮平一水合物晶体进行干燥处理来制备氯氮平无水物晶体颗粒。首先对氯氮平一水合物在25℃下,进行搅拌存在的氮气吹扫干燥,干燥时间大约为5小时。关闭氮气并对体系进行抽真空处理,同时将温度提升到40℃,保持搅拌,干燥3-5小时,同时取样测其含水量,当含水量小于0.2%时停止干燥。所得的产物即氯氮平无水物晶体。
[0031]
在一些实施例中,所述有机溶剂为乙醇、丙酮或n,n-二甲基甲酰胺。
[0032]
在一些实施例中,所述添加剂为乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素或木质素磺酸钠。
[0033]
在一些实施例中,添加剂的添加量为50-1000ppm。
[0034]
在一些实施例中,结晶的温度范围为10-25℃。
[0035]
在一些实施例中,所述过滤采用0.45微米有机滤膜。
[0036]
在一些实施例中,对氯氮平一水合物采用氮吹干燥和真空干燥。
[0037]
在一些实施例中,所述氮吹干燥在25~26℃下,干燥时间5~6小时,再在真空条件下,于40~42℃干燥3~5小时。
[0038]
下面结合具体的实施例,对本发明做进一步的详细说明,应该指出,所述具体实施例是对本发明的解释而不是限定。
[0039]
以下实施例中,总体质量收率=氯氮平无水物晶体质量/氯氮平粗品质量
×
100%。
[0040]
实施例1:
[0041]
步骤一:将10g氯氮平粗品溶解于100g无水乙醇中,溶解温度为20℃。溶解后将溶液过0.45微米有机滤膜,滤液转移到250ml夹套结晶器中待用。将0.02g羟丙基甲基纤维素溶解于100g去离子水中,溶解完全后过0.45微米水性滤膜待用。盛有氯氮平溶液的夹套结晶器温度控制在20℃,开启搅拌,搅拌速度设定为150rpm,用蠕动泵向氯氮平母液中以1ml/min的速度滴加添加剂的水溶液。完全滴加结束后,恒温搅拌杨晶1小时。将产物经过过滤、去离子水洗涤后,在40℃真空烘箱中干燥4小时,即得到氯氮平一水合物晶体。晶体颗粒分散度高,平均粒度在150微米左右,质量收率为82%。
[0042]
步骤二:将步骤一得到的氯氮平一水合物放置于200ml夹套干燥器中,在25℃氮气吹扫干燥5小时,期间开启搅拌器,搅拌转速200rpm。之后将温度升高至40℃,关闭氮气,体系抽真空,在此条件下干燥5小时。期间采用水分仪实时测定固体的含水量,待含水量小于0.5%时收料。得到的产物即为颗粒分散度高的氯氮平无水物晶体。产物平均粒度为120微米,总体质量收率为80%,堆密度0.62g/ml,振实密度0.71g/ml,休止角40
°
,其中,氯氮平显微镜图如图1所示,氯氮平xrd图谱如图2所示,氯氮平粒度分布图如图3所示,氯氮平tg图如图4所示,氯氮平sem图如图5所示,药学指标均合格。
[0043]
实施例2:
[0044]
步骤一:将10g氯氮平粗品溶解于100g丙酮中,溶解温度为20℃。溶解后将溶液过0.45微米有机滤膜,滤液转移到250ml夹套结晶器中待用。将0.02g乙基纤维素溶解于100g去离子水中,溶解完全后过0.45微米水性滤膜待用。盛有氯氮平溶液的夹套结晶器温度控制在20℃,开启搅拌,搅拌速度设定为150rpm,用蠕动泵向氯氮平母液中以1ml/min的速度
滴加添加剂的水溶液。完全滴加结束后,恒温搅拌杨晶1小时。将产物经过过滤、去离子水洗涤后,在40℃真空烘箱中干燥4小时,即得到氯氮平一水合物晶体。晶体颗粒分散度高,平均粒度在160微米左右,质量收率为85%。
[0045]
步骤二:将步骤一得到的氯氮平一水合物放置于200ml夹套干燥器中,在25℃氮气吹扫干燥5小时,期间开启搅拌器,搅拌转速200rpm。之后将温度升高至40℃,关闭氮气,体系抽真空,在此条件下干燥5小时。期间采用水分仪实时测定固体的含水量,待含水量小于0.5%时收料。得到的产物即为颗粒分散度高的氯氮平无水物晶体。产物平均粒度为140微米,总体质量收率为82%,堆密度0.65g/ml,振实密度0.75g/ml,休止角38
°
,药学指标均合格。
[0046]
实施例3:
[0047]
步骤一:将10g氯氮平粗品溶解于100gn,n-二甲基甲酰胺中,溶解温度为20℃。溶解后将溶液过0.45微米有机滤膜,滤液转移到250ml夹套结晶器中待用。将0.02g木质素磺酸钠溶解于100g去离子水中,溶解完全后过0.45微米水性滤膜待用。盛有氯氮平溶液的夹套结晶器温度控制在20℃,开启搅拌,搅拌速度设定为150rpm,用蠕动泵向氯氮平母液中以1ml/min的速度滴加添加剂的水溶液。完全滴加结束后,恒温搅拌杨晶1小时。将产物经过过滤、去离子水洗涤后,在40℃真空烘箱中干燥4小时,即得到氯氮平一水合物晶体。晶体颗粒分散度高,平均粒度在155微米左右,质量收率为86%。
[0048]
步骤二:将步骤一得到的氯氮平一水合物放置于200ml夹套干燥器中,在25℃氮气吹扫干燥5小时,期间开启搅拌器,搅拌转速200rpm。之后将温度升高至40℃,关闭氮气,体系抽真空,在此条件下干燥5小时。期间采用水分仪实时测定固体的含水量,待含水量小于0.5%时收料。得到的产物即为颗粒分散度高的氯氮平无水物晶体。产物平均粒度为140微米,总体质量收率为83%,堆密度0.60g/ml,振实密度0.70g/ml,休止角38
°
,药学指标均合格。
[0049]
最后应该说明的是,以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。