一种3
×
flag标签融合表达载体的制备方法及其应用
技术领域
1.本发明属于分子生物学技术领域,具体涉及一种3
×
flag标签融合表达载体的制备方法及其应用。
背景技术:
2.flag是一段由8个氨基酸残基(n-dykddddk-c)组成的多肽,大约1012da。在蛋白质表达、蛋白质互作等研究中,可以通过基因工程技术手段将所要研究的目的基因和flag多肽序列连接起来,flag多肽可以连接在目的蛋白的c端或n端,然后将“目的基因-flag”融合后的表达载体转入细菌、酵母、动物或者植物细胞中。一方面,因为flag标签的分子量很小,所以不会遮盖住融合蛋白中的蛋白表位和结构域,也不容易改变融合蛋白的功能、定位、分泌和运输等。flag标签中的四个连续的天冬氨酸(dddd)带负电荷,可以让整个标签具有天然的亲水特性,使得flag标签很容易定位在融合蛋白的表面,便于利用抗体来检测。另一方面,目前flag标签蛋白的抗体已经商业化大规模生产,很多品牌如国外品牌sigma、abcam和国产品牌abmart、翌圣等的flag标签抗体已广泛用于“目的基因-flag”融合蛋白的定量、定位或者蛋白互作研究,检测手段有免疫荧光,免疫印记等。
3.目前植物中flag标签融合表达载体应用较广泛的多为pcambia1300框架,该载体框架较大,达10kb以上。该框架多用于农杆菌介导的烟草植株的转化。尽管应用这种方法可以得到大量表达的“目的基因-flag”融合蛋白,但是烟草育苗周期长,农杆菌介导的转化步骤繁杂。另外,烟草系统无法模拟不同植物本身环境,而对蛋白功能和蛋白互作的研究,特别是利用“目的基因-flag”融合蛋白载体系统寻找目的基因在植物体内的互作蛋白时,要求“目的基因-flag”融合载体要在所研究的目标植物中表达,此时农杆菌介导的转化方法在其它植物如拟南芥、水稻、玉米中的应用受到阻碍。
4.peg介导的植物原生质体转化是目前应用较多的融合载体表达系统,但是pcambia1300框架太大,转化效率低,且pcambia1300框架拷贝数低,无法满足原生质体转化系统的大量质粒要求。因此,在实际分子生物学研究中急需一种能用于植物原生质体高效转化和表达的flag标签融合表达载体。
技术实现要素:
5.本发明所要解决的技术问题在于针对上述现有技术的不足,提供一种3
×
flag标签融合表达载体的制备方法及其应用,该方法制备的3
×
flag标签融合表达载体pproto-3
×
flag框架较小,全长4088bp,适用于植物原生质体转化。
6.为解决上述技术问题,本发明采用的技术方案是:一种3
×
flag标签融合表达载体的制备方法,该方法为:
7.s1、用kpni/xhoi双酶切pgl3 basic载体,在温度为37℃的条件下反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下4.8kb大小的条带,用dna凝胶回收试剂盒回收产物,得到kpni/xhoi双酶切pgl3 basic载体的回收产物;
8.s2、以prgeb32bar-cas9质粒为模板,以特异性引物f1和特异性引物r1为引物进行pcr反应克隆2
×
35s启动子,pcr扩增后,将pcr扩增产物用1%琼脂糖凝胶分离,切下677bp大小的条带,用dna凝胶回收试剂盒回收pcr产物,得到2
×
35s启动子pcr回收产物;pcr反应体系为:2
×
pfu master mix 10μl,特异性引物f11μl、特异性引物r11μl、prgeb32bar-cas9质粒1μl、灭菌超纯水补至20μl;pcr扩增的反应条件为:98℃预变性10min;98℃变性10s,55℃退火10s,68℃延伸40s,扩增40个循环;68℃延伸10min;所述特异性引物f1的核苷酸序列如seq id no:1所示;所述特异性引物r1核苷酸序列如seq id no:2所示;
9.s3、利用同源重组方法将s2中得到的2
×
35s启动子pcr回收产物中的2
×
35s启动子连接至s1中得到的kpni/xhoi双酶切pgl3 basic载体的回收产物中pgl3 basic载体的kpni/xhoi位点之间,在温度为50℃的条件下连接反应25min,得到pgl-35s连接产物,将所述pgl-35s连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s;
10.s4、将s3中得到的中间载体pgl-35s用xhoi/sali双酶切,在温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.67kb大小的条带,用dna凝胶回收试剂盒回收产物,得到xhoi/sali双酶切中间载体pgl-35s的回收产物;
11.s5、人工合成多克隆位点mcs正向引物和多克隆位点mcs反向引物,将所述多克隆位点mcs正向引物和所述多克隆位点mcs反向引物在95℃的条件下退火5min,得到mcs退火引物;所述多克隆位点mcs正向引物的核苷酸序列如seq id no:3所示;所述多克隆位点mcs反向引物的核苷酸序列如seq id no:4所示;
12.s6、将s5中得到的mcs退火引物t4连接至s4中得到的xhoi/sali双酶切中间载体pgl-35s的回收产物中pgl-35s的xhoi/sali位点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs;
13.s7、克隆nos ter序列:以pcambia1302质粒为模板,以特异性引物f2和特异性引物r2为引物进行pcr反应克隆nos ter序列,pcr扩增后,将pcr扩增产物用1.5%琼脂糖凝胶分离,切下251bp大小的条带,用dna凝胶回收试剂盒回收pcr产物,得到nos ter序列pcr回收产物;pcr反应体系为:2
×
pfu master mix 10μl,特异性引物f21μl、特异性引物r21μl、pcambia1302质粒1μl、灭菌超纯水补至20μl;pcr扩增的反应条件为:98℃预变性10min;98℃变性10s,55℃退火10s,68℃延伸20s,扩增40个循环;68℃延伸10min;所述特异性引物f2的核苷酸序列如seq id no:5所示;所述特异性引物r2核苷酸序列如seq id no:6所示;
14.s8、将s6中得到的中间载体pgl-35s-mcs用ecori/sali双酶切,在温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.72kb大小的条带,用dna凝胶回收试剂盒回收产物,得到ecori/sali双酶切中间载体pgl-35s-mcs的回收产物;
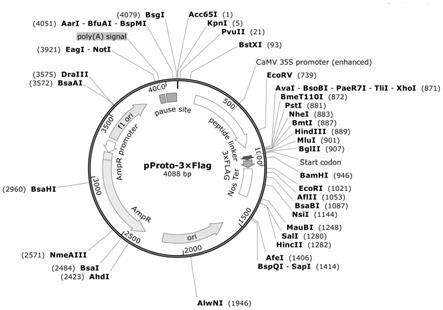
15.s9、利用同源重组方法将s7中得到的nos ter序列pcr回收产物中的nos ter序列连接至s8中得到ecori/sali双酶切中间载体pgl-35s-mcs的回收产物中的pgl-35s-mcs的ecori/sali位点之间,在温度为50℃的条件下连接反应25min,得到pgl-35s-mcs-nos连接产物,将所述pgl-35s-mcs-nos连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs-nos;
16.s10、将s9中得到的中间载体pgl-35s-mcs-nos用bglii/bamhi双酶切,温度为37℃
的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.97kb大小的条带,用dna凝胶回收试剂盒回收产物,得到bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物;
17.s11、体外人工合成连接肽linker正向引物和连接肽linker反向引物,将所述连接肽linker正向引物和所述连接肽linker反向引物在温度为95℃的条件下退火5min,得到变性退火的连接肽linker引物;所述连接肽linker正向引物的核苷酸序列如seq id no:7所示;所述连接肽linker反向引物的核苷酸序列如seq id no:8所示;
18.s12、将s11中得到的变性退火的连接肽linker引物t4连接至s10中得到的bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物中pgl-35s-mcs-nos的bglii/bamhi位点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs-linker-nos连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs-linker-nos;
19.s13、将s12中得到的中间载体pgl-35s-mcs-linker-nos用bamhi/ecori双酶切,温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下4.01kb大小的条带,用dna凝胶回收试剂盒回收产物,得到bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物;
20.s14、合成3
×
flag标签正向引物和3
×
flag标签反向引物,将所述3
×
flag标签正向引物和所述3
×
flag标签反向引物在温度为95℃的条件下退火5min,得到3
×
flag标签退火引物;所述3
×
flag标签正向引物的核苷酸序列如seq id no:9所示;所述3
×
flag标签反向引物的核苷酸序列如seq id no:10所示;
21.s15、将s14中得到的3
×
flag标签退火引物t4连接至s13中得到的bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物中pgl-35s-mcs-linker-nos的bamhi/ecori位点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs-linker-nos连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到3
×
flag标签融合表达载体,命名为3
×
flag标签融合表达载体pproto-3
×
flag;所述3
×
flag标签融合表达载体pproto-3
×
flag的核苷酸如seq id no:11所示。
22.优选地,s1中用kpni/xhoi双酶切pgl3 basic载体的酶切体系为:kpni酶1μl、xhoi酶1μl、cutsmart buffer 5μl、pgl3 basic载体10μl、灭菌超纯水补至50μl;
23.s3中所述连接反应的反应体系为:2
×
hieffclone enzyme premix 5μl、s1中得到的kpni/xhoi双酶切pgl3 basic载体的回收产物2μl、s2中得到的2
×
35s启动子pcr回收产物1μl、灭菌超纯水补至10μl;
24.s4中所述酶切反应的体系为:酶切体系为:sali酶1μl、xhoi酶1μl、cutsmart buffer 5μl、s3中得到的中间载体pgl-35s 10μl、灭菌超纯水补至50μl;
25.s5中所述退火的反应体系为:所述多克隆位点mcs正向引物10μl、所述多克隆位点mcs反向引物10μl、灭菌超纯水补至100μl;
26.s6中所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s4中所述xhoi/sali双酶切中间载体pgl-35s的回收产物2μl、s5中所述mcs退火引物1μl、灭菌超纯水补至10μl;
27.s8中所述酶切反应的体系为:ecori酶1μl、sali酶1μl、cutsmart buffer 5μl、s6
中得到的中间载体pgl-35s-mcs 10μl、灭菌超纯水补至50μl;
28.s9中所述连接反应的反应体系为:2
×
hieffclone enzyme premix 5μl、s6中得到的ecori/sali双酶切中间载体pgl-35s-mcs的回收产物2μl、s7中得到的nos ter序列pcr回收产物1μl、灭菌超纯水补至10μl;
29.s10中所述酶切反应的体系为:bglii酶1μl、bamhi酶1μl、cutsmart buffer 5μl、s9中得到的中间载体pgl-35s-mcs-nos 10μl、灭菌超纯水补至50μl;
30.s11中所述退火的反应体系为:所述连接肽linker正向引物10μl、所述连接肽linker反向引物10μl、灭菌超纯水补至100μl;
31.s12中所述连接反应的体系为:所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s10中所述bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物2μl、s11中所述变性退火的连接肽linker引物1μl、灭菌超纯水补至10μl;
32.s13中所述酶切反应的体系为:bamhi酶1μl、ecori酶1μl、cutsmart buffer 5μl、s12中得到的中间载体pgl-35s-mcs-linker-nos 10μl、灭菌超纯水补至50μl;
33.s15所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s13中所述bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物2μl、s14中所述3
×
flag标签退火引物1μl、灭菌超纯水补至10μl。
34.本发明还提供了上述制备的3
×
flag标签融合表达载体的应用,所述3
×
flag标签融合表达载体用于植物原生质体的转化。
35.优选地,所述3
×
flag标签融合表达载体用于目的蛋白在植物原生质体中的表达。
36.优选地,所述植物原生质体包括拟南芥原生质体、玉米原生质体。
37.本发明与现有技术相比具有以下优点:
38.1、本发明的3
×
flag标签融合表达载体pproto-3
×
flag框架较小,全长4088bp,适用于植物原生质体转化。且载体框架上含有高拷贝数的细菌的复制子(ori),容易得到大量纯化的质粒用于原生质体转化。本发明的3
×
flag标签融合表达载体pproto-3
×
flag中多克隆位点前方具有2
×
35s启动子(camv 35s promoter en3
×
flagnced),相比于单一的camv35s promoter,它能够更强的驱动“目的基因-flag”在植物原生质体表达。本发明的3
×
flag标签融合表达载体将三个flag标签串联在一起,这样的设计能增加flag标签蛋白和抗体的结合,进而增加检测的敏感度。
39.本发明的3
×
flag标签融合表达载体pproto-3
×
flag中3
×
flag标签和多克隆位点前有一段连接肽linker,富含甘氨酸的柔性单元,可有助于保持目的蛋白的蛋白表位和结构域。
40.下面结合附图和实施例对本发明作进一步详细说明。
附图说明
41.图1是本发明的实施例1的pgl3 basic载体的物理图谱。
42.图2是本发明的实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag的物理谱图。
43.图3是本发明的实施例2的pproto-yfp-3
×
flag融合表达载体转化至玉米原生质体后的yfp发光图。
44.图4是本发明的实施例2的pproto-yfp-3
×
flag融合表达载体转化至玉米原生质体后的western blotting图。
45.图5是本发明的实施例2的pproto-atpif4-3
×
flag融合表达载体转化至拟南芥原生质体的western blot图。
46.图6是本发明的实施例2的pproto-yfp-3
×
flag融合表达载体转化至拟南芥原生质体后的yfp发光图。
具体实施方式
47.实施例1
48.本实施例的3
×
flag标签融合表达载体的制备方法,该方法为:
49.s1、用kpni/xhoi双酶切pgl3 basic载体(pgl3 basic载体的物理图谱为图1),在温度为37℃的条件下反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下4.8kb大小的条带,用dna凝胶回收试剂盒回收产物,得到kpni/xhoi双酶切pgl3 basic载体的回收产物;用kpni/xhoi双酶切pgl3 basic载体的酶切体系为:kpni酶1μl、xhoi酶1μl、cutsmart buffer5μl、pgl3 basic载体10μl、灭菌超纯水补至50μl;
50.s2、以prgeb32bar-cas9质粒(mk791524.1)为模板,以特异性引物f1和特异性引物r1为引物进行pcr反应克隆2
×
35s启动子,pcr扩增后,将pcr扩增产物用1%琼脂糖凝胶分离,切下677bp大小的条带,用dna凝胶回收试剂盒回收pcr产物,得到2
×
35s启动子pcr回收产物;pcr反应体系为:2
×
pfu master mix 10μl,特异性引物f11μl、特异性引物r11μl、prgeb32bar-cas9质粒1μl、灭菌超纯水补至20μl;pcr扩增的反应条件为:98℃预变性10min;98℃变性10s,55℃退火10s,68℃延伸40s,扩增40个循环;68℃延伸10min;所述特异性引物f1的核苷酸序列如seq id no:1所示;所述特异性引物r1核苷酸序列如seq id no:2所示;
51.s3、利用同源重组方法将s2中得到的2
×
35s启动子pcr回收产物中的2
×
35s启动子连接至s1中得到的kpni/xhoi双酶切pgl3 basic载体的回收产物中pgl3 basic载体的kpni/xhoi位点之间,在温度为50℃的条件下连接反应25min,得到pgl-35s连接产物,将所述pgl-35s连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s;所述连接反应的反应体系为:2
×
hieffclone enzyme premix5μl、s1中得到的kpni/xhoi双酶切pgl3 basic载体的回收产物2μl、s2中得到的2
×
35s启动子pcr回收产物1μl、灭菌超纯水补至10μl;
52.s4、将s3中得到的中间载体pgl-35s用xhoi/sali双酶切,在温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.67kb大小的条带,用dna凝胶回收试剂盒回收产物,得到xhoi/sali双酶切中间载体pgl-35s的回收产物;所述酶切反应的体系为:酶切体系为:sali酶1μl、xhoi酶1μl、cutsmart buffer 5μl、s3中得到的中间载体pgl-35s 10μl、灭菌超纯水补至50μl;
53.s5、人工合成多克隆位点mcs正向引物和多克隆位点mcs反向引物,将所述多克隆位点mcs正向引物和所述多克隆位点mcs反向引物在95℃的条件下退火5min,得到mcs退火引物;所述多克隆位点mcs正向引物的核苷酸序列如seq id no:3所示;所述多克隆位点mcs反向引物的核苷酸序列如seq id no:4所示;所述退火的反应体系为:所述多克隆位点mcs
正向引物10μl、所述多克隆位点mcs反向引物10μl、灭菌超纯水补至100μl;
54.s6、将s5中得到的mcs退火引物t4连接至s4中得到的xhoi/sali双酶切中间载体pgl-35s的回收产物中pgl-35s的xhoi/sali位点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs;所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s4中所述xhoi/sali双酶切中间载体pgl-35s的回收产物2μl、s5中所述mcs退火引物1μl、灭菌超纯水补至10μl;
55.s7、克隆nos ter序列:以pcambia1302质粒为模板,以特异性引物f2和特异性引物r2为引物进行pcr反应克隆nos ter序列,pcr扩增后,将pcr扩增产物用1.5%琼脂糖凝胶分离,切下251bp大小的条带,用dna凝胶回收试剂盒回收pcr产物,得到nos ter序列pcr回收产物;pcr反应体系为:2
×
pfu master mix 10μl,特异性引物f21μl、特异性引物r21μl、pcambia1302质粒1μl、灭菌超纯水补至20μl;pcr扩增的反应条件为:98℃预变性10min;98℃变性10s,55℃退火10s,68℃延伸20s,扩增40个循环;68℃延伸10min;所述特异性引物f2的核苷酸序列如seq id no:5所示;所述特异性引物r2核苷酸序列如seq id no:6所示;
56.s8、将s6中得到的中间载体pgl-35s-mcs用ecori/sali双酶切,在温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.72kb大小的条带,用dna凝胶回收试剂盒回收产物,得到ecori/sali双酶切中间载体pgl-35s-mcs的回收产物;所述酶切反应的体系为:ecori酶1μl、sali酶1μl、cutsmart buffer 5μl、s6中得到的中间载体pgl-35s-mcs 10μl、灭菌超纯水补至50μl;
57.s9、利用同源重组方法将s7中得到的nos ter序列pcr回收产物中的nos ter序列连接至s8中得到ecori/sali双酶切中间载体pgl-35s-mcs的回收产物中的pgl-35s-mcs的ecori/sali位点之间,在温度为50℃的条件下连接反应25min,得到pgl-35s-mcs-nos连接产物,将所述pgl-35s-mcs-nos连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs-nos;所述连接反应的反应体系为:2
×
hieff clone enzyme premix 5μl、s6中得到的ecori/sali双酶切中间载体pgl-35s-mcs的回收产物2μl、s7中得到的nos ter序列pcr回收产物1μl、灭菌超纯水补至10μl;
58.s10、将s9中得到的中间载体pgl-35s-mcs-nos用bglii/bamhi双酶切,温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下3.97kb大小的条带,用dna凝胶回收试剂盒回收产物,得到bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物;所述酶切反应的体系为:bglii酶1μl、bamhi酶1μl、cutsmart buffer 5μl、s9中得到的中间载体pgl-35s-mcs-nos 10μl、灭菌超纯水补至50μl;
59.s11、体外人工合成连接肽linker正向引物和连接肽linker反向引物,将所述连接肽linker正向引物和所述连接肽linker反向引物在温度为95℃的条件下退火5min,得到变性退火的连接肽linker引物;所述连接肽linker正向引物的核苷酸序列如seq id no:7所示;所述连接肽linker反向引物的核苷酸序列如seq id no:8所示;所述退火的反应体系为:所述连接肽linker正向引物10μl、所述连接肽linker反向引物10μl、灭菌超纯水补至100μl;
60.s12、将s11中得到的变性退火的连接肽linker引物t4连接至s10中得到的bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物中pgl-35s-mcs-nos的bglii/bamhi位
点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs-linker-nos连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到中间载体pgl-35s-mcs-linker-nos;所述连接反应的体系为:所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s10中所述bglii/bamhi双酶切中间载体pgl-35s-mcs-nos的回收产物2μl、s11中所述变性退火的连接肽linker引物1μl、灭菌超纯水补至10μl;
61.s13、将s12中得到的中间载体pgl-35s-mcs-linker-nos用bamhi/ecori双酶切,温度为37℃的条件下酶切反应6h后,将酶切产物用1%琼脂糖凝胶分离后,切下4.01kb大小的条带,用dna凝胶回收试剂盒回收产物,得到bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物;所述酶切反应的体系为:bamhi酶1μl、ecori酶1μl、cutsmart buffer 5μl、s12中得到的中间载体pgl-35s-mcs-linker-nos 10μl、灭菌超纯水补至50μl;
62.s14、合成3
×
flag标签正向引物和3
×
flag标签反向引物,将所述3
×
flag标签正向引物和所述3
×
flag标签反向引物在温度为95℃的条件下退火5min,得到3
×
flag标签退火引物;所述3
×
flag标签正向引物的核苷酸序列如seq id no:9所示;所述3
×
flag标签反向引物的核苷酸序列如seq id no:10所示;
63.s15、将s14中得到的3
×
flag标签退火引物t4连接至s13中得到的bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物中pgl-35s-mcs-linker-nos的bamhi/ecori位点之间,在温度为22℃的条件下连接反应1h,得到pgl-35s-mcs-linker-nos连接产物;将所述pgl-35s-mcs连接产物转化到大肠杆菌中,扩增繁殖,用质粒提取试剂盒提取后,得到3
×
flag标签融合表达载体,命名为3
×
flag标签融合表达载体pproto-3
×
flag(3
×
flag标签融合表达载体pproto-3
×
flag的物理谱图为图2);所述3
×
flag标签融合表达载体pproto-3
×
flag的核苷酸如seq id no:11所示;所述连接反应的体系为:t4连接缓冲液1μl、t4连接酶1μl、s13中所述bamhi/ecori双酶切中间载体pgl-35s-mcs-linker-nos的回收产物2μl、s14中所述3
×
flag标签退火引物1μl、灭菌超纯水补至10μl。
64.实施例2
65.本实施例为实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag的应用,所述3
×
flag标签融合表达载体pproto-3
×
flag用于植物原生质体的转化;用于目的蛋白在植物原生质体中的表达;所述植物原生质体包括拟南芥原生质体、玉米原生质体。
66.(1)克隆增强型黄色荧光蛋白(yfp),通过同源重组将yfp连接至实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag的xhoi/hindiii之间,得到pproto-yfp-3
×
flag融合表达载体。
67.利用peg介导的方法将pproto-yfp-3
×
flag融合表达载体转化至玉米原生质体,大量表达yfp-3
×
flag融合蛋白。
68.将转化后的玉米原生质体在激光共聚焦显微镜488nm激发光下观察发光情况,如图3,可见视野里基本所有的细胞都能发光,图3中左侧为黄色荧光蛋白yfp发光图片(yfp),右侧为明场的细胞图片(bright field)(左右两图共用同一标尺)。
69.将转化后的玉米原生质体在25℃黑暗培养16h后,150g离心3min收集原生质体,用终浓度1
×
loading buffer重悬原生质体,95c,5min,sds-page胶分离,western blotting检测,使用3
×
flag抗体为abbkine(a02011),yfp-3
×
flag融合蛋白的检测结果如图4,箭头指示黄色荧光蛋白yfp的western blot条带。说明实施例1制备的3
×
flag标签融合表达载
体pproto-3
×
flag可以成功用于目的蛋白在植物原生质体中的表达和检测。
70.(2)克隆拟南芥atpif4基因的cds(at2g43010),通过同源重组将atpif4连接至实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag的xhoi/hindiii之间,得到pproto-atpif4-3
×
flag融合表达载体。利用peg介导的方法将pproto-atpif4-3
×
flag融合表达载体转化至拟南芥原生质体。
71.25℃黑暗培养16h后,150g离心3min收集原生质体,用裂解缓冲液重悬细胞后,加入3
×
flag beads(sigma,m8823),4℃孵育24h,用1
×
tbs清洗beads 3次后,加入40μl裂解缓冲液和终浓度1
×
loading buf fer,95℃,5min,sds-page胶分离,western blotting检测,使用3
×
flag抗体为abbkine(a02011)。结果如图5,箭头指示atpif4-3
×
flag融合蛋白的input(总蛋白样)的western blot条带(wb)和ip免疫沉淀条带(ip),说明实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag可以成功用于目的蛋白在所研究的植物原生质体中表达,并可利用3
×
flag beads进行目的蛋白的免疫沉淀。
72.(3)克隆增强型黄色荧光蛋白(yfp),通过同源重组将yfp连接至实施例1制备的3
×
flag标签融合表达载体pproto-3
×
flag的xhoi/hindiii之间,得到pproto-yfp-3
×
flag融合表达载体。利用peg介导的方法将pproto-yfp-3
×
flag融合表达载体转化至拟南芥原生质体。
73.将转化后的拟南原生质体在激光共聚焦显微镜488nm激发光下观察发光情况,如图6,可见视野里基本所有的细胞都能发光,图6中左侧为黄色荧光蛋白yfp发光图片(yfp),右侧为明场的细胞图片(bright field)(左右两图共用同一标尺)。
74.以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制。凡是根据发明技术实质对以上实施例所作的任何简单修改、变更以及等效变化,均仍属于本发明技术方案的保护范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。