1.本发明涉及嵌合分子、药物组合物、靶核酸的切割方法以及靶核酸切割用或诊断用试剂盒。
2.本技术基于2019年7月24日在日本技术的日本特愿2019-136414号主张优先权,并将其内容引用于此。
背景技术:
3.近年来,核酸药物与抗体药物同样作为下一代分子靶向药物而受到关注。特别地,核酸药物还涉及不能用抗体药物治疗的疾病。另外,具有通过化学合成可以比较廉价地提供等许多优点,与抗体药物同样,作为后低分子药物的地位也逐渐确立。
4.核酸药物中报告了多种药物策略,例如反义核酸(aso)以与疾病进展相关的信使rna(mrna)、微rna(mirna)、小干扰rna(sirna)等为靶标,选择性地识别碱基序列,形成复合物,从而抑制靶rna的功能,发挥治疗效果。为了这些核酸药物有效地发挥药效,需要1)高体内稳定性,2)对靶核酸的高特异性和复合物稳定性,对天然型dna/rna进行化学修饰的寡核酸/人工寡核酸的开发正在积极地研究。
5.虽然迄今为止报道了优异的修饰或人工核酸,但指出由与具有与靶核酸类似序列的非靶核酸的结合引起的副作用(狭义的脱靶效果)、不依赖于靶核酸识别的核酸药物特有的毒性(广义的脱靶效果)的发现是面向实用化而应解决的课题,其降低、改善正在世界范围内进行研究。
6.作为克服广义脱靶效果的方法论,提出了降低核酸药物的给药量的方案。但是,如果减少给药量,当然会引起与靶rna形成复合物的量降低,不能期待有效的药效发挥。具体地,使用报道的细胞内导入量以亚nm水平为极限的aso,不仅在表达量为亚μm水平的mrna为靶标的系统内,在以nm-pm水平的细胞内表达量发挥功能的mirna为靶标的系统也报道了反馈机制,并指出基于靶rna与aso的1:1复合物形成的药效发挥策略不能预期足够的治疗效果。
7.作为本课题的解决方法,利用少量的aso就可以像催化剂那样切割靶rna的活用rnaseh的具有催化剂样功能的核酸药物受到关注(非专利文献1)。
8.现有技术文件
9.非专利文献
10.非专利文献1:liang,x.et al.,mol.ther.2017,25,2075
技术实现要素:
11.本发明的目的在于提供可以以低浓度抑制靶核酸的功能、可以抑制脱靶效应的嵌合分子、包含所述嵌合分子的药物组合物、使用所述嵌合分子的靶核酸的切割方法、以及包含所述嵌合分子的靶核酸切割用或诊断用试剂盒。
12.本发明的发明人认为,增加代谢更新率对于提高rna酶切割靶rna的效率是有效的。从该观点出发,通过着眼于rna切割后的解离过程,提出了有助于构建可从切割后靶rna的复合物迅速解离的寡核酸系的设计方法,并成功进行实证实验。发现利用本方法,则可以以低浓度抑制靶核酸的功能,可以抑制脱靶效果,从而完成了本发明。
13.本发明包括以下方式。
14.[1]一种嵌合分子,其为对靶核酸具有结合能力的第一核酸或其衍生物、以及对所述靶核酸具有结合能力且主链骨架为阴离子性的第二核酸或其衍生物融合而成。
[0015]
[2]根据[1]所述的嵌合分子,其中,所述第一核酸或其衍生物的主链骨架为中性或阳离子性。
[0016]
[3]根据[2]所述的嵌合分子,其中,所述第一核酸或其衍生物的主链骨架为酰胺骨架。
[0017]
[4]根据[1]-[3]中任意一项所述的嵌合分子,其中,所述第二核酸或其衍生物的主链骨架为糖-磷酸骨架。
[0018]
[5]根据[1]-[4]中任意一项所述的嵌合分子,其中,所述第一核酸或其衍生物与所述第二核酸或其衍生物的5’末端融合。
[0019]
[6]根据[1]-[4]中任意一项所述的嵌合分子,其中,所述第一核酸或其衍生物与所述第二核酸或其衍生物的3’末端融合。
[0020]
[7]根据[1]-[6]中任意一项所述的嵌合分子,其中,由所述嵌合分子和与所述嵌合分子结合的所述靶核酸构成的复合物与核酸酶特异性结合。
[0021]
[8]根据[7]所述的嵌合分子,其中,所述核酸酶在所述第一核酸或其衍生物与所述第二核酸或其衍生物的融合部分切割所述靶核酸。
[0022]
[9]根据[8]所述的嵌合分子,其中,用所述核酸酶切割后的两个靶核酸片段的熔解温度tm为38℃以下。
[0023]
[10]根据[7]-[9]中任意一项所述的嵌合分子,其中,所述核酸酶为核糖核酸酶h。
[0024]
[11]根据[1]-[10]中任意一项所述的嵌合分子,其中,所述第一核酸或其衍生物为肽核酸或肽核糖核酸或者它们的衍生物。
[0025]
[12]根据[1]-[11]中任意一项所述的嵌合分子,其中,所述第二核酸或其衍生物为dna。
[0026]
[13]根据[1]-[12]中任意一项所述的嵌合分子,其中,所述靶核酸为rna或dna。
[0027]
[14]一种药物组合物,其以[1]-[13]中任意一项所述的嵌合分子作为有效成分。
[0028]
[15]根据[14]所述的药物组合物,其中,所述药物组合物为癌症或缺血性脑疾病。
[0029]
[16]一种靶核酸的切割方法,其使用[1]-[13]中任意一项所述的嵌合分子和核酸酶来切割靶核酸。
[0030]
[17]根据[16]所述的靶核酸的切割方法,其中,所述核酸酶为核糖核酸酶h。
[0031]
[18]一种靶核酸切割用或诊断用试剂盒,其包含[1]-[13]中任意一项所述的嵌合分子和核酸酶。
[0032]
[19]根据[18]所述的试剂盒,其中,所述核酸酶为核糖核酸酶h。
[0033]
根据本发明,能够提供可以以低浓度抑制靶核酸的功能、可以抑制脱靶效应的嵌合分子、包含所述嵌合分子的药物组合物、使用所述嵌合分子的靶核酸的切割方法、以及包
含所述嵌合分子的靶核酸切割用或诊断用试剂盒。
附图说明
[0034]
图1是使用与各10倍量的5’末端用荧光色素标记的rna混合的体系,用聚丙烯酰胺凝胶电泳分析prpd-rna复合物的由rnaseh产生的切割物的图。泳道1示出rna1的ladder和碱基序列,泳道2示出在dna1和rna1的复合物中添加60u/μl的rnaseh的情况下、泳道3示出在dna1和rna1的复合物中添加6u/μl的rnaseh的情况下、泳道4示出在prpd1和rna1的复合物中添加60u/μl的rnaseh的情况下、泳道5示出在prpd1和rna1的复合物中添加6u/μl的rnaseh的情况下由rnaseh产生的rna1切割物的聚丙烯酰胺凝胶电泳图案。
[0035]
图2a是示出在dna1-rna1复合物的情况下rnaseh的rna1切割位点的图。
[0036]
图2b是示出在prpd1-rna1复合物的情况下rnaseh的rna1切割位点的图。
[0037]
图3是使用与各10倍量的5’末端用荧光色素标记的rna混合的体系,用聚丙烯酰胺凝胶电泳分析dna1、prpd4、prpd1或pd2与rna1的复合物的由rnaseh产生的切割物的图。泳道1示出分别添加rna1(对照)、dna1、prpd4、prpd1、pd2的情况下由rnaseh产生的rna1切割物的聚丙烯酰胺凝胶电泳图案。泳道6示出rna1的ladder和碱基序列。
[0038]
图4是使用与各10倍量的5’末端用荧光色素标记的rna混合的体系,用聚丙烯酰胺凝胶电泳分析dna2、prpd7或pd1与rna2的复合物的由rnaseh产生的切割物的图。泳道1示出rna2的ladder和碱基序列,泳道2-5分别示出添加rna2(对照)、dna2、prpd7、pd1的情况下由rnaseh产生的rna2切割物的聚丙烯酰胺凝胶电泳图案。
[0039]
图5是使用与各10倍量的5’末端用荧光色素标记的rna混合的体系,用聚丙烯酰胺凝胶电泳分析dprp1或prpd5与rna1的复合物的由rnaseh产生的切割物的图。泳道1示出rna1的ladder和核苷酸序列,泳道2-8分别示出添加rna1(对照)、prpd5(10nm)、prpd5(1nm)、prpd5(0.1nm)、dprp1(10nm)、dprp1(1nm)和dprp1(0.1nm)的情况下由rnaseh产生的rna1切割物的聚丙烯酰胺凝胶电泳图案。
[0040]
图6是示出rnaseh对dprp1-rna1复合物的rna切割位点的图。
[0041]
图7示出在无细胞翻译系统中prpd2和dna2的反义活性。泳道1-3示出在rnaseh不存在下的结果,泳道4-6示出在rnaseh存在下的结果。泳道1和4示出添加对照的情况,泳道2和5示出添加dna2的情况,泳道3和6示出添加prpd2的情况。
[0042]
图8示出在无细胞翻译系统中prpd5和dprp2的反义活性。泳道1、3和5示出在rnaseh不存在下的结果,泳道2、4和6示出在rnaseh存在下的结果。泳道1和2示出添加对照的情况,泳道3和4示出添加prpd5的情况,泳道5和6示出添加dprp2的情况。
具体实施方式
[0043]
[嵌合分子]
[0044]
本发明的嵌合分子为对靶核酸具有结合能力的第一核酸或其衍生物、以及对所述靶核酸具有结合能力且主链骨架为阴离子性的第二核酸或其衍生物融合而成。
[0045]
本发明中,作为核酸的衍生物,没有特别限制,可以举出与核酸结合的碱基部为尿嘧啶、胞嘧啶、胸腺嘧啶、腺嘌呤、鸟嘌呤或者嘌呤环或嘧啶环的卤化衍生物、脱氨基衍生物、或具有硫原子代替各核酸碱基的氧原子的衍生物等。
[0046]
所述对靶核酸具有结合能力的第一核酸或其衍生物优选主链骨架为中性或阳离子性,更优选为中性。作为所述中性的主链骨架,没有特别限制,例如可以举出酰胺骨架。作为所述具有酰胺骨架的核酸或其衍生物,可以举出例如肽核酸或其衍生物(以下也称为peptide nucleic acid;pna)、肽核糖核酸或其衍生物(peptide ribonucleic acid;以下也称为prna)、prna和pna的组合(以下也称为pna/prna)等。pna/prna中,pna中prna的结合位点可为pna的任意位点,也可结合到pna的中间。例如可以为pna-prna-pna这样的组合。
[0047]
作为所述阳离子性的主链骨架,没有特别限制,例如可以举出亚氨基骨架、磷酸酰胺或亚膦酰胺骨架等。具有所述各骨架的核酸或其衍生物例如有吗啉代基型核酸等。
[0048]
作为对所述靶核酸具有结合能力且主链骨架为阴离子性的第二核酸或其衍生物的主链骨架,没有特别限制,但优选糖-磷酸骨架。作为具有糖-磷酸骨架的核酸或其衍生物,例如,可以举出核糖核酸(ribonucleic acid;以下也称为rna)、脱氧核糖核酸(deoxyribonucleic acid;以下也称为dna)等。
[0049]
作为所述靶核酸,只要是具有本发明的嵌合分子能够结合的靶序列的核酸或其衍生物即可,没有特别限制,但优选rna或dna,当本发明的嵌合分子用作药物组合物时,所述靶核酸优选编码导致用所述药物组合物治疗的疾病的蛋白质的rna或dna。
[0050]
所述第一核酸或其衍生物可以融合至所述第二核酸或其衍生物的3’末端和5’末端中的任意一者。
[0051]
作为本发明的所述第一核酸或其衍生物与所述第二核酸或其衍生物融合的嵌合分子,例如可以举出作为所述第一核酸或其衍生物的pna与作为所述第二核酸或其衍生物的dna的融合物(以下也称为pna-dna嵌合分子),作为所述第一核酸或其衍生物的pna/prna与作为所述第二核酸或其衍生物的dna的融合物等。作为作为所述第一核酸或其衍生物的pna/prna与作为所述第二核酸或其衍生物的dna的融合物,可以举出pna/prna与dna的5’侧融合的嵌合分子(以下也称为pna/prna-dna嵌合分子、prpd)、pna/prna与dna的3’侧融合的嵌合分子(以下也称为dna-pna/prna嵌合分子、dprp)等。
[0052]
本发明的嵌合分子可以如下制备。
[0053]
所述第一核酸为pna且所述第二核酸为dna的pna-dna嵌合分子,例如可以如下制备。
[0054]
(pna低聚物的合成)
[0055]
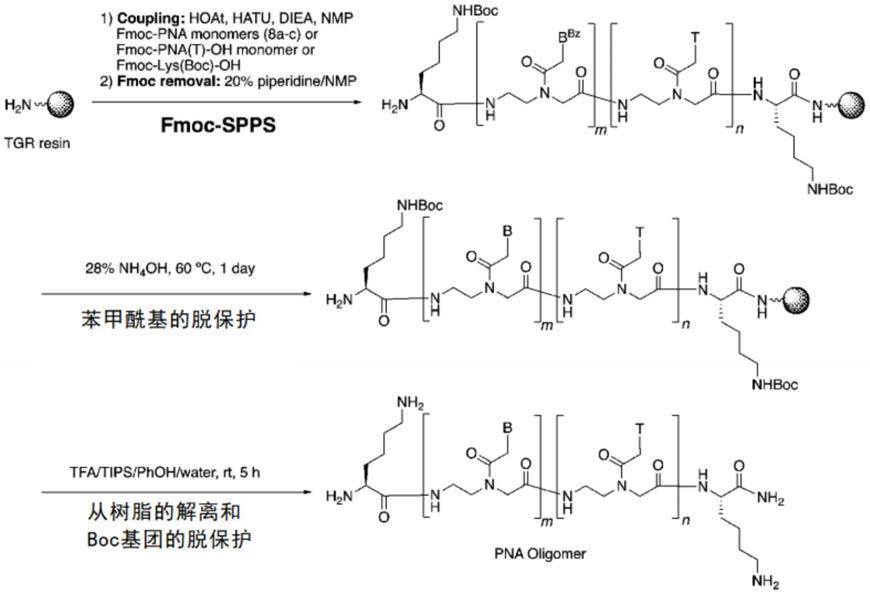
pna低聚物可以通过公知的固相肽合成方法合成。例如,可以使用苯甲酰基保护fmocpna单体,通过fmoc-固相肽合成法(solid phase peptide synthesis;以下也称为fomc-spps法),通过下述工序进行合成。
[0056]
[化学式1]
[0057][0058]
在上述中,为了提高水溶性和抑制酰基转移反应而导入n末端或c末端的赖氨酸残基。合成可以使用novasyn(注册商标)tgr树脂等,通过公知的方法进行。苯甲酰基保护fmoc-pna单体的缩合反应例如可以通过在含有fmoc-pna(bz)-oh(10当量)、hatu(1-[bis(dimethylamino)methyliumyl]-1h-1,2,3-triazolo[4,5-b]pyridine-3-oxide hexafluorophosphate;10当量)、hoat(1-hydroxy-7-azabenzotriazole;10当量)和diea(n,n-diisopropylethylamine;20当量)的nmp(n-methylpyrrolidone)溶液中处理30分钟来进行。fmoc基团可通过用含有20%哌啶的nmp溶液处理20分钟而除去。重复这些偶联、fmoc基团的除去步骤,直到达到目标序列。苯甲酰基的脱保护可以通过用28%氨水在60℃下处理来进行。最后,赖氨酸残基的boc基团的解离和脱保护用tfa(trifluoroacetic acid)/tips(triisopropylsilane)/苯酚/水(94:3:1:2)的混合物处理来进行。得到的粗产物可以通过反相hplc纯化,纯化产物可以通过esi-tof-ms鉴定。
[0059]
(pna-dna嵌合分子的合成)
[0060]
pna-dna嵌合分子可通过将通过公知方法合成的苯甲酰基保护fmoc单体应用于目标序列,例如可以如下合成。
[0061]
[化学式2]
[0062][0063]
pna分子的n末端用甘氨酸修饰以抑制酰基转移反应。使用与腺嘌呤残基结合的cpg树脂,通过dna/rna自动合成仪从dna分子的构建开始合成dna。
[0064]
dna分子可以通过使用5-bmt作为激活剂的通常的亚磷酰胺法延伸。使用通过公知方法制备的5
’‑
mmtr(单甲氧基三苯甲基)氨基脱氧核苷酸亚磷酰胺,将5
’‑
氨基修饰的脱氧核苷酸衍生物导入dna分子的5’末端。在通过dna合成仪导入5’氨基修饰的衍生物后,通过用3%tca/dcm溶液处理15分钟,除去dna5’末端的mmtr基团。接着,为了通过酰胺键键合pna,使用dna的5’氨基并通过fmoc-spps法使其延伸。
[0065]
使用苯甲酰基保护fmocpna的缩合反应在含有fmoc-pna(bz)-oh(10当量)、hatu(10当量)、hoat(10当量)和diea(20当量)的nmp溶液中进行30分钟。树脂上未反应的氨基的乙酰基封端可以在25%乙酸酐/dcm下进行。fmoc基团可以通过20%哌啶/nmp处理除去。重复这些偶联、乙酰封端和fmoc除去步骤,直到获得目标序列。最后,苯甲酰基和dna分子的氰乙基的解离和脱保护通过用28%氨处理60分钟来进行。所得粗产物可通过反相hplc纯化,且纯化物可通过maldi-tof-ms鉴定。
[0066]
(pna/prna-dna嵌合分子;prpd的合成)
[0067]
pna/prna与dna的5’侧融合的prpd的合成可以通过与所述pna-dna嵌合分子的合成同样的方法,例如通过下述工序进行。
[0068]
[化学式3]
[0069][0070]
可以基于fmoc-spps法将rpna单体导入pna分子的特定位点。prna单体(fmoc-γprna-oh)例如可以通过以下工序制备。
[0071]
[化学式4]
[0072][0073]
粗产物经反相hplc纯化,纯化物可以通过maldi-tof-ms鉴定。
[0074]
(dna-pna/prna嵌合分子;dprp的合成)
[0075]
pna/prna与dna的3’末端融合的dprp的合成可以通过例如fmoc-spps法,使用dna合成仪,通过以下工序进行。
[0076]
[化学式5]
[0077][0078]
合成通过使用1-(3-二甲基氨基丙基)-3-乙基碳化二亚胺盐酸盐(edc
·
hcl)和4-二甲基氨基吡啶的dmf(n,n-二甲基甲酰胺,n,n-dimethylformamide)溶液的缩合试剂,在novasyn(注册商标)tga树脂上导入fmoc-gly-oh来开始。
[0079]
合成优选使用fmoc-gly功能性tga树脂以fmoc-spps法进行。pna/prna分子可以通过fmoc-spps法使用苯甲酰基保护fmoc-pna和fmoc-γprna单体延长来进行合成。偶联条件可以与pna-dna嵌合分子的条件相同。fmoc-spps完成后,prna分子的2’,3
’‑
羟基用乙酰基保护。除去n末端的fmoc基团后,将树脂用dna合成仪,通过公知的亚磷酰胺法,从以亚磷酰胺键结合的pna/prna分子的n末端氨基延伸dna分子。最后,用28%氨进行解离和脱保护。得到的粗产物可通过反相hplc纯化,纯化物可以通过maldi-tof-ms鉴定。
[0080]
(对核酸酶的作用)
[0081]
本发明的嵌合分子和与所述嵌合分子结合的所述靶核酸形成的复合物与核酸酶特异性结合。作为所述核酸酶,优选使用核糖核酸酶h,但不限于此,只要与所述复合物特异性结合即可,对核糖核酸酶的种类没有特别限制。
[0082]
所述核酸酶具有识别所述第二核酸或其衍生物的位点(以下也称为核酸识别位点)和切割靶核酸的核酸酶活性位点。所述核酸酶的所述核酸识别位点由于具有由碱性氨基酸残基构成的通道结构,从而与具有阴离子骨架的所述第二核酸或其衍生物结合。当所
述第二核酸或其衍生物与所述核酸识别位点结合时,与所述第二核酸或其衍生物融合的第一核酸或其衍生物被吸引至所述核酸酶的切割活性位点,从而在所述第一核酸或其衍生物与所述第二核酸或其衍生物的融合(接合)位点处选择性地切割靶核酸。
[0083]
本发明的嵌合分子能够在所述第一核酸或其衍生物与所述第二核酸或其衍生物之间的融合(接合)位点切割与本发明的嵌合分子结合的靶序列,因此通过设计具有作为靶核酸的切割位点的所述诱导位点的嵌合分子,从而可以在目标位点切割靶核酸。
[0084]
在本发明的嵌合分子中,用所述核酸酶切割后的两个靶核酸片段的熔解温度(tm)优选为体温以下,例如可以为38℃以下,也可以为37℃以下、30℃以下、25℃以下。通过使所述两个靶核酸片段的tm为体温以下,可以使由所述核酸酶在一个位置切割而产生的两个靶核酸片段迅速地从核酸酶解离。另一方面,在核酸酶切割靶核酸后,本发明的嵌合分子从核酸酶迅速解离,因此本发明的嵌合分子迅速用于下一次靶核酸切割,能够实现核酸酶对靶核酸切割的高效更新。因此,本发明的嵌合分子能够以低浓度切割靶核酸,从而能够减轻脱靶效果。
[0085]
[药物组合物]
[0086]
本发明的药物组合物含有本发明的嵌合分子作为有效成分。
[0087]
在本发明中,作为药物组合物,没有特别限制,可以举出治疗由本发明的嵌合分子靶向的靶核酸编码的蛋白质引起的疾病的药物组合物等,例如抗肿瘤剂、缺血性脑疾病治疗剂等。本发明的药物组合物可以通过内源性核酸酶切割靶核酸,抑制靶核酸编码的蛋白质的表达,因此能够治疗由所述靶核酸编码的蛋白质引起的疾病。
[0088]
作为所述靶核酸编码的蛋白质,只要是成为本发明的药物组合物的靶标的蛋白质,则在原理上均可,例如,作为本发明的药物组合物的靶标的蛋白质的例子,可以举出作为特发性肺纤维化的治疗靶标的tgfβ、作为癌症的治疗靶标的p53以及癌基因ras、作为老年性黄斑变性症的治疗靶标的vegf165等。
[0089]
作为所述疾病,没有特别限制,可以举出癌症、缺血性脑病、老年性黄斑变性、家族性高胆固醇血症、肌营养不良、痴呆症、nash、肝硬化、特发性肺纤维化、肝病、自身免疫性疾病、肾病、造血系统疾病、特应性皮肤病、银屑病等。
[0090]
本发明的药物组合物可以是各种形式,例如液剂(例如注射剂)、分散剂、悬浮剂、片剂、丸剂、粉末剂、栓剂等。优选的方案为注射剂,优选非经口(例如静脉内、经皮、腹膜内、肌内)给药。
[0091]
作为本发明的药物组合物,不仅可以根据上述剂型以单剂使用,还可以选择与不同组成的核酸药物或低分子药物、抗体药物等的合剂等,给药时使用的剂型或输送系统最合适的剂型或输送系统。另外,可以选择将本发明的药物组合物直接或通过修饰而封入胶束内、或与抗体药物等结合的形态等各种给药方法。本发明的药物组合物根据疾病的种类或靶核酸的种类等而不同,例如可以为0.025-50mg/kg,优选为0.1-50mg/kg,更优选为0.1-25mg/kg,进一步优选为0.1-10mg/kg或0.1-3mg/kg,但不限于此。
[0092]
[靶核酸的切割方法和靶核酸切割用或诊断用试剂盒]
[0093]
本发明的靶核酸的切割方法使用本发明的嵌合分子和核酸酶。作为核酸酶,可以举出上述的核酸酶。通过本发明的靶核酸的切割方法,可以特异性切割靶序列,因此,除了序列选择性切割靶核酸、基因组编辑工具以外,该方法还可以用于诊断由靶核酸编码的蛋
白质引起的疾病。
[0094]
本发明的靶核酸切割用或诊断用试剂盒包含本发明的嵌合分子和核酸酶。作为核酸酶,可以举出上述的核酸酶。本发明的靶核酸切割用试剂盒中,除了本发明的嵌合分子和核酸酶以外,还可以包含靶核酸片段检测所需的其他构成要素,例如丙烯酰胺凝胶、反应缓冲液、反应容器等。通过本发明的靶核酸切割用试剂盒,可以特异性切割靶核酸,因此可以用作序列选择性切割靶核酸、基因组编辑工具。另外,本发明的诊断用试剂盒可以用于诊断由本发明的嵌合分子靶向的靶核酸编码的蛋白质引起的疾病。作为由本发明的嵌合分子靶向的靶核酸编码的蛋白质,可以举出上述的蛋白质。
[0095]
本发明的诊断试剂盒可以通过检测例如受试者的生物样品中的核酸是否可被本发明的诊断试剂盒切割来确定受试者的生物样品中是否包含靶核酸,因此可以诊断受试者是否患有由本发明的嵌合分子靶向的靶核酸编码的蛋白质引起的疾病。作为所述生物样品,没有特别限制,例如可以举出血液、唾液、尿、脊髓液、骨髓液、胸腔积液、腹水、关节液、泪液、玻璃体液、玻璃体液和淋巴液等。
[0096]
另外,通过将本发明的诊断用试剂盒的构成要素导入体内,也可以应用于在体外检测由于体内环境或目标分子的存在与否、浓度等而产生的检测探针分子的结构变化或物理化学变化,通过在外部检测由从体外照射或暴露的光或磁、超声波、放射线等引起的能量变化来高灵敏度地测量,作为图像而可视化的体外诊断。
[0097]
实施例
[0098]
下面,示出实施例对本发明进行更详细的说明,但本发明并不限定于以下的实施例。
[0099]
[实施例]pna-dna嵌合分子的制备
[0100]
pna-dna嵌合分子通过dna自动合成和使用cpg树脂的fmoc-spps法合成。
[0101]
合成通过自动dna合成仪从dna分子的构建开始。为了合成载有2
’‑
脱氧腺苷残基的cpg树脂dna,将与腺嘌呤残基结合的cpg树脂(1μm级)导入dna合成仪,以dna分子作为激活剂,通过使用5-bmt的通常的亚磷酰胺法延长。亚磷酰胺溶液的浓度为乙腈中70-78mm。用按照公知的方法制备的5
’‑
mmtrnh脱氧胞苷亚磷酰胺,将5
’‑
氨基修饰脱氧胞苷衍生物导入dna分子的5’末端。使用最终的三苯甲基“on”工序,通过dna合成仪导入5
’‑
氨基修饰衍生物后,从dna合成仪上移除树脂并转移到用于fmoc-spps工序的反应柱。通过用3%tca/dcm溶液处理15分钟,除去dna的5’末端的mmtr基团。
[0102]
然后,使用dna的5’氨基作为酰胺键的基础,通过fmoc-spps法使pna延伸。为了使表面的氨基成为碱性,用20%哌啶/nmp处理树脂,过滤,用nmp清洗5次以上。使用苯甲酰基保护fmocpna单体的缩合反应,通过使用含有fmoc-pna(bz)-oh(10当量)、hatu(10当量)、hoat(10当量)和diea(20当量)的nmp溶液(300μl)的处理进行。在室温下反应30分钟,同时不时强烈搅拌,然后除去溶液,用nmp洗涤树脂3次以上。
[0103]
然后,将在nmp中的20%哌啶溶液添加到树脂中,不时搅拌20分钟以除去fmoc基团。fmoc基团除去完成后,用nmp清洗树脂5次以上。重复缩合反应和fmoc除去工序,直至构建目标pna-dna嵌合分子的序列。
[0104]
最后,通过与上述相同的缩合条件导入fmoc-gly-oh,以抑制pna残基的n-末端氨基内的分子酰基转移。fmoc-spps工序完成后,通过将树脂在28%氨溶液中在60℃下处理16
小时,除去所有保护基,从树脂中分离。然后,通过过滤除去树脂,浓缩滤液。使用cosmosil 5c
18-ar-ii柱(nacalai,10φ
×
250mm),在3ml/min流速下,使用溶剂a:0.1m teaa缓冲液(ph7.5)、溶剂b:0.1m teaa(ph7.5)/乙腈(1:1,v/v)在5%至70%线性梯度、30分钟的条件下,使用反相hplc纯化粗产物。
[0105]
通过上述方法制备下述表1所示的2种pna-dna嵌合分子(pd1、pd2)。
[0106]
[表1]
[0107][0108]
小写字母表示pna碱基,大写字母表示dna碱基,
hn
t、
hn
c表示5
’‑
氨基取代dna碱基。
[0109]
[实施例2]prpd的制备
[0110]
作为在dna的5’末端侧融合pna/prna的dna-pna/prna嵌合的prpd是通过使用dna自动合成仪和cpg树脂的fmoc工序的组合来合成的。
[0111]
合成通过自动dna合成从dna分子的构建开始。将载有2’脱氧核苷残基的cpg树脂(1μm级)导入dna合成仪,通过使用5-bmt作为激活剂的通常的亚磷酰胺法延长dna分子的长度。亚磷酰胺溶液的浓度为乙腈中70-78mm。用按照公知的方法制备的5
’‑
mmtrnh脱氧胞苷亚磷酰胺,将5’氨基修饰的脱氧核苷酸衍生物导入dna分子的5’末端。
[0112]
使用最终的三苯甲基“on”工序,通过dna合成仪导入5
’‑
氨基修饰衍生物后,从dna合成仪上移除树脂并转移到用于fmoc-spps工序的反应柱。通过用3%tca/dcm溶液处理15分钟,除去dna的5’末端的mmtr基团。
[0113]
然后,使用dna的5’氨基作为酰胺键的基础,通过fmoc-spps法使pna延伸。为了使表面的氨基成为碱性,用20%哌啶/nmp处理树脂,过滤,用nmp清洗5次以上。
[0114]
使用苯甲酰基保护fmocpna单体的缩合反应,通过使用含有fmoc-pna(bz)-oh(10当量)、hatu(10当量)、hoat(10当量)和diea(20当量)的nmp溶液(300μl)的处理进行。在室温下反应30分钟,同时不时强烈搅拌,然后除去溶液,用nmp洗涤树脂3次以上。
[0115]
然后,将在nmp中的20%哌啶溶液添加到树脂中,并不时搅拌20分钟以除去fmoc基团。fmoc基团除去完成后,用nmp清洗树脂5次以上。重复缩合反应和fmoc除去工序,直至构建目标pna-dna嵌合序列。最后,通过与上述相同的缩合条件导入fmoc-gly-oh或fmoc-lys(fmoc)-oh,以抑制pna残基的n-末端氨基内的分子酰基转移。
[0116]
fmoc-spps工序完成后,通过将树脂在28%氨溶液中在60℃下处理16小时,除去所有保护基,从树脂中分离。
[0117]
然后,通过过滤除去树脂,浓缩滤液。使用cosmosil 5c
18-ar-ii柱(nacalai,10φ
×
250mm),在3ml/min流速下,使用溶剂a:0.1m teaa缓冲液(ph7.5)、溶剂b:0.1m teaa(ph7.5)/乙腈(1:1,v/v)在5%至70%线性梯度、30分钟的条件下,使用反相hplc纯化粗产物。
[0118]
通过上述方法制备表2所示的7种prpd(prpd1~prpd7)。
[0119]
[表2]
[0120][0121]
斜体字母表示prna碱基,小写字母表示pna碱基,大写字母表示dna碱基,
nh
t、
nh
c表示5
’‑
氨基取代dna碱基。
[0122]
[实施例3]dprp的制备
[0123]
将fmoc-gly-oh(30mg,100μmol)和edc
·
hcl(19mg,100μmol)溶于dmf(1.0ml)中,并将溶液在冰冷却下搅拌35分钟。除去冰冷却,将在dmf和dmap(1.2mg,10μmol)中的novasyn(注册商标)tga树脂(42mg,10μmol的羟基)加入dmf和dmap(1.2mg,10μmol)至溶液中,并将得到的混合物在室温下搅拌1.5小时。过滤树脂并用nmp、dcm/乙醇(1:1,v/v)和dcm连续洗涤。
[0124]
在充分洗涤树脂后,将1.0ml苯甲酸酐(113mg,0.5mmol)的20%吡啶/nmp溶液加入到树脂中,并在不时振荡的同时将混合物放置在室温下。放置1小时后,用nmp和dcm充分洗涤树脂。在用乙醇洗涤后,在减压下在干燥器中干燥树脂,得到fmoc-gly功能性tga树脂(41mg,94%)。通过定量uv(301nm)测量由20%哌啶/nmp处理而游离的fmoc-gly-oh来计算fmoc-gly-oh的导入量。
[0125]
将如上得到的fmoc-gly功能性tga树脂(1μmol级)用nmp洗涤,并在室温下用20%哌啶/nmp处理20分钟以除去fmoc保护基。在用nmp充分洗涤后,将树脂加入到fmoc-pna(bz)-oh或fmoc-γrpna-oh(10当量)、hoat(10当量)、hatu(10当量)和diea(20当量)的nmp溶液(300μl)中,并将混合物一边不时振荡,一边在室温下静置30分钟,并将该偶联工序重复两次。在用nmp和dcm洗涤后,在室温下用25%乙酸酐/dcm处理树脂10分钟。在上述偶联工序后,将树脂用dcm洗涤,用nmp洗涤。接着,在室温下通过20%哌啶/nmp处理20分钟除去fmoc。使用相同的fmoc除去、偶联和封端工序延伸pna-prna链。在最后的偶联工序后,用nmp和dcm充分地洗涤树脂,并用吡啶洗涤。然后,将乙酸酐/dcm(2:3,v/v)添加到树脂中,并使其在室温下静置2小时,同时不时振荡以保护prna分子的2
’3’
羟基。
[0126]
在n-末端fmoc保护基被除去后,将树脂转移到用于自动dna/rna合成仪的空柱中,并将含有树脂的柱子安装在dna/rna合成仪上。用5
’‑
o-dmtr-3
’‑
o-(2-氰基乙基)亚磷酰胺构建块(構築
ブロック
),通过通常的亚磷酰胺方法延伸dna链。在链组装之后,用28%氨水在60℃下处理树脂18小时,从而使dprp脱保护并解离。然后,通过膜滤器进行过滤,除去树脂。将滤液在真空下部分浓缩,并使用反相hplc在5c
18-ar-ii柱(10
×
250nm)上纯化,得到dprp。
[0127]
通过上述方法,制备了表3所示的两种dprp(dprp1、dprp2)。
[0128]
[表3]
[0129][0130]
斜体字母表示prna碱基,小写字母表示pna碱基,大写字母表示dna碱基,
hn
t、
hn
c表示5
’‑
氨基取代dna碱基。
[0131]
[实施例4]rnase活性的测定
[0132]
如下研究由rnase切割prpd
·
rna复合物的rna切割活性。
[0133]
将作为靶标的、碱基序列具有序列编号12所示碱基序列的5
’‑
fam标记rna(5
’‑
fam-gaaucuuauagucuugca-3’;rna1)与实施例2中得到的prpd1的0.1当量混合。将混合物退火以形成prpd-rna复合物。
[0134]
然后,向混合物中加入60u/μl的rnaseh,在37℃下使其反应,切割rna1。反应30分钟后,加入7m尿酸tris盐酸缓冲液停止反应,然后在70℃下进行10分钟rnaseh的灭活。作为prpd1的对照,对具有配对部分的序列编号13所示碱基序列的dna1(5
’‑
tgcaagactataagattc-3’)进行相同的处理。切割后的rna片段通过用20%分解聚丙烯酰胺凝胶电泳(分解page)、荧光图像分析仪的可视化和定量进行分析。其结果示于图1和表4。
[0135]
[表4]
[0136][0137]
如图1和表4所示,在dna1使用60u/μl的rnaseh的情况下,54%的rna1被切割。与之相对,在prpd1的情况下,超过99%的rna1被切割,这意味着rnaseh的活性饱和。因此,将rnaseh的量减少至6u/μl,进行相同rna的切割实验。
[0138]
结果如图1和表4所示,dna1的情况下rna1的切割程度减少至12%,而prpd1的情况下rna1的切割维持在90%以上。其结果表明,rnaseh与dna1
·
rna1复合物相比,在prpd1
·
rna1复合物中显示出8倍高的rna1切割活性。
[0139]
prpd1的切割位点如图2b所示,为pna-dna酰胺键附近的一个位点。与之相对,在dna1的情况下,如图2a所示,在多个位点被切割。rnaseh结合的最小dna碱基长为5-6个碱基,这对应于rnaseh的dna结合通道间的距离。因此,prpd1的dna分子由7个dna核酸碱基组成,可以通过导入一个切割位点的rnaseh的dna结合通道正确识别。使用5-6个碱基对的dna-rna复合物可以在几个位点连接。这诱导多个位点的rna切割。基于这些,rna切割的促进解释如下。
[0140]
rnaseh对靶rna不具有碱基序列选择性,因此dna1的情况下,与靶rna的复合物的切割位点是各种各样的,一次切割不能引起复合物解离的可能性高,不能从aso-切割rna-rnaseh复合物解离切割rna,dna1的更新次数降低。另一方面,可以认为在作为本发明的嵌合分子的prpd1的情况下,能够实现在靶rna的一个位点、特别是切割后的复合物的稳定性均为体温(37℃)以下的序列前后的切割,通过一次切割,复合物能够高效解离,与其他未切
割的靶rna结合,从而能够飞跃性提高更新数。这是通过使用了实际测定或热力学参数的最接近碱基法算出切割后的rna1
·
dna1复合物的切割后的两个片段与aso的复合物的tm而证实的。
[0141]
如图2a所示,在dna1的情况下,在不同位点(a~e)非特异性地观察到切割。计算切割后的复合物的稳定性,在位点a和位点d的切割中,与dna1的切割后复合物分别为tm=35.3℃、37.2℃。这与体温(37℃)大致相同,可以预想到即使在切割后复合物也难以解离。即使在其它位点进行切割,与切割片段的复合物的tm也多为37℃以上。一部分切割片段(位点b、c)在切割后的rna片段与dna1的复合物的tm虽然也有37℃以下的情况,但其生成概率不高。因此,通常复合物解离需要多次rnaseh切割,引起rnaseh切割的更新数的抑制。
[0142]
与此相反,prpd1在pna-dna连接处显示出选择性的一种rna切割,这与rna切割前相比大大降低了rna1
·
prpd1复合物的温度稳定性。因此,通过一次rnaseh切割,复合物解离,aso能够与未被立即切割的靶rna形成复合物,结果rnaseh切割更新数得到促进,在该实验中,所有rna1被切割。为了确认这一点,计算切割后的rna-prpd1复合物的tm。
[0143]
如图2b所示,主要的切割位点是位点a中的一个。由此,生成的片段形成与prpd1的复合物不同类型的复合物,如prpd1与dna分子的复合物(切割后的rna
·
dna复合物)、prpd1与pna/prna分子的复合物(切割后的rna
·
pna/prna复合物)。上述切割后的rna
·
dna复合物的温度稳定性通过最近邻动态参数算出,结果tm=13.2℃。上述切割后的rna
·
pna/prna复合物的温度稳定性经uv温度熔化分析为tm=22.3℃。据报道pna在生理高盐浓度条件下与dna/rna形成不稳定复合物。由此认为,切割后的rna
·
pna/prna复合物的这种低温度稳定性是合理的。
[0144]
由以上可知,在pna-dna融合部中选择性的一个位点上的rna1的切割生成具有体温(37℃)以下温度稳定性的rna片段,被识别为prpd1的有效更新。
[0145]
接下来,研究了与rnaseh活性相关的prna向嵌合物的导入效果的影响。在dna1、prpd4、prpd1或pd2(10nm)存在下,使rna1(1μm)与rnaseh(0.6u/μl)在37℃下反应30分钟。其结果示于图3和表5。
[0146]
[表5]
[0147][0148]
在dna1的情况下,rna1的切割量非常小。与之相对,在10nm的prpd4和prpd1存在下,观察到超过90%的rna1切割。对于pd2,与prpd4和prpd1相比,也显示相同水平的rnaseh活性。
[0149]
使用16merrna,对与rnaseh活性相关、导入嵌合物的prna的效果进行了研究,该16merrna使用了具有序列编号14所示碱基序列的rna2(5
’‑
fam-uaagaaggagauauac-3’)。在具有序列编号15所示碱基序列的dna2(5
’‑
gtatatctccttctta-3’)、prpd7或pd1(10nm)存在
下,使rna2(1μm)与rnaseh(6
×
10-3u/μl)在37℃下反应30分钟。其结果示于图4和表6。
[0150]
[表6]
[0151][0152]
在dna2的情况下,52%的rna2被切割。在prpd7的存在下,观察到76%的rna2切割,其rnaseh活性是dna2的1.5倍。与之相对,在pd1的情况下,rna2切割量为40%,其比prpd7低1.9倍。其结果,prna向嵌合分子的导入使得能够促进rnaseh活性,这被认为是由于与靶rna形成a型复合物的rna选择性结合性。
[0153]
然后,研究了通过dprp1的rnaseh对靶rna1的切割活性,并与prpd5的活性进行比较。预期dprp在3’末端诱导rna切割。将rna1与1/100、1/1000或1/10000当量的dprp1或prpd5混合,分别退火以形成复合物。然后,加入6
×
10-3
u/μl的rnaseh,在37℃下反应30分钟。其结果示于图5和表7。
[0154]
[表7]
[0155][0156]
如图5所示,dprp1的rna在距rna的3’末端3-4个碱基的位点处被切割。主要的切割位点如图6所示。10nm dprp1的切割活性为43%,其比dprp5的切割活性低1.7倍(73%)。dprp1的这种低切割活性被认为是由于限制性的rna1切割位点。在prpd5的情况下,切割位点是pna-dna连接处的一个点。因此,通过生成具有37℃以下的低温度稳定性的rna1,实现了rna1的有效解离和prpd5的有效更新。
[0157]
与之相对,在dprp1的情况下,在rna1的3’末端附近观察到切割,因此在切割后产生长rna片段,其保持37℃以上的高温度稳定性。因此,dprp1的更新效率与prpd5相比降低。
[0158]
[实施例5]rnaseh的rna切割反应的动力学参数
[0159]
为了研究prpd5的高效rna切割,进行rnaseh的rna切割动力学分析。
[0160]
在过量prpd1
·
rna1(1、2、5、10μm)的存在下,使rnaseh(1.5
×
10-3
u/μl=4.1nm)在pseudo-first-order反应条件下反应,并绘制在各prpd1
·
rna1浓度下切割的rna1量的时间曲线。通过使用得到的各底物浓度的初始速度,进行lineweaver-burk作图。对于dprp1
·
rna1也进行相同的实验。将由lineweaver-burk作图得到的动力学参数总结于表8中。dna
·
rna复合物的参数参照大塚等报告的结果进行提取。其结果示于表8。
[0161]
[表8]
[0162][0163]
如表8所示,prpd1
·
rna1复合物与rnaseh的rna切割反应的k
cat
值与dna
·
rna复合物相比为1.9倍左右。该结果显示,rnaseh与对应的dna
·
rna复合物相比,prpd1
·
rna1复合物切割效率有若干提高,但与图3所示的靶rna切割实验相比,其提高率明显较低。rnaseh显示出与prpd1
·
rna1复合物的高切割活性,rnaseh与prpd1
·
rna1复合物的结合的michaelis-menten乘数(km)比dna
·
rna复合物大。这表明prpd1
·
rna1复合物与rnaseh的结合活性低于dna
·
rna复合物。根据k
cat
和km值之间的关系认为,rnaseh的识别性对prpd1的高rna切割活性几乎没有影响。另外,prpd1
·
rna1复合物的vmax值比dna
·
rna复合物大4倍,但该值比由凝胶电泳推定的值小(例如,根据图1,观察到约8倍高的rna切割活性(泳道2与4的比较)。由这些结果表明,rnaseh与底物(prpd1-rna1复合物)的结合不是控速步骤,切割后的prpd1-rna1复合物从rnaseh解离的工序对于rnaseh的有效rna切割活性是重要的。
[0164]
在dprp1的情况下,rnaseh对rna的切割活性k
cat
值比dna
·
rna复合物小。该结果显示,dprp1与对应的dna相比显示出低的切割活性。rna切割的vmax值为dna
·
rna复合物的1.7倍以上,而km值为dna
·
rna复合物的5.2倍以上。
[0165]
与该dna
·
rna复合物相比,高km值可以认为是由天然dna骨架置换为pna-prna骨架引起的rnaseh识别性降低所致。然而,dprp1
·
rna1复合物的km值低于prpd1-rna1,这表明与prpd1
·
rna1复合物相比,dprp1
·
rna1复合物的rnaseh识别性增强。在dprp1
·
rna1和prpd1
·
rna1之间km值的不同支持了通过rnaseh的dprp1
·
rna1复合物的识别位点不同于prpd1-rna1复合物的识别位点。
[0166]
图5所示的切割反应也支持识别位点不同。prpd与dprp之间的不同rnaseh活性被认为不仅依赖于rna切割位点,而且依赖于rnaseh的识别性和rnaseh的催化切割活性(k
cat
/km)。该k
cat
/km值可以通过依赖于rnaseh的rna切割反应的动力学分析的底物浓度来确定。如上表8中所示,prpd和dprp的k
cat
/km值为几乎相同的量级(prpd
·
rna:1.67
×
106s-1
m-1
,dprp
·
rna:1.97
×
106s-1
m-1
)。由此可以认为嵌合分子的更新效率主要受rna切割位点的控制。由此可知,rnaseh的切割位点对于促进rna切割活性和嵌合的更新效率是重要的。
[0167]
[实施例6]无细胞翻译系统中的反义活性
[0168]
为了确认对内源性rna切割的应用,使用体外无细胞翻译系统研究了prpd的蛋白质合成抑制活性(反义活性)。作为用于该研究的aso的prpd2和dna2为16mer,其包含5
’‑
gaagga-3’,这是用于荧光素酶报告测定的renilla荧光素酶基因上游的shine-dalgarno(sd)序列的互补序列。相对于靶mrna,添加1/10当量的aso(prpd2或dna2)。在rnaseh存在和不存在下进行反应,以评价rnaseh的rna切割活性。其结果示于图7。
[0169]
如图7所示,在rnaseh不存在下,在dna2的情况下几乎观察不到反义活性。通过添加rnaseh,略微观察到dna2的抑制活性(12%)。与之相对,在prpd2的情况下,在rnaseh的不存在下,观察到9%的反义活性,该反义活性略高于dna1。prpd2的反义活性通过rnaseh添加显著地增加至91%。比rnaseh不存在下的化学计量抑制水平低表明抑制仅由1:1复合物形成引起。仅添加1/10当量的反义寡核苷酸,prpd2的情况下,确认到10%以上的蛋白质表达
的抑制。这表明prpd2通过rnaseh活性起催化作用。该结果表明prpd能够有效地切割靶mrna,并利用rnaseh活性抑制蛋白质生产。该结果与rna片段的凝胶电泳分析一致。因此,认为研究rna的切割位点对设计用于反义活性的更有效的aso是非常重要的。
[0170]
然后,用pursystem classic ii研究无细胞翻译系统中prpd5和dprp2的反义活性。实验在rnaseh不存在和存在下进行。其结果示于图8。
[0171]
相对于编码renilla荧光素酶基因的靶mrna,添加1/10当量的prpd5和dprp2。在prpd5的情况下,通过rnaseh添加将反义活性提高至75%。与之相对,dprp2通过添加rnaseh促进反义活性为12%,与prpd5相比低6倍。
[0172]
工业实用性
[0173]
根据本发明,能够提供可以以低浓度抑制靶核酸的功能、可以抑制脱靶效应的嵌合分子、包含所述嵌合分子的药物组合物、使用所述嵌合分子的靶核酸的切割方法、以及包含所述嵌合分子的靶核酸切割用或诊断用试剂盒。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。