1.本发明属于生物工程领域,具体涉及一种葛根素酸酯衍生物及其制备方法与应用。
背景技术:
2.葛根素,又称为8-β-d-葡萄吡喃糖-4',7-二羟基异黄酮,是野葛根中主要活性成分之一。葛根素化学结构中的b环受到了吡喃环羰基的影响,从而形成了一个大的共轭系统。同时,在7,4’位点上的两个酚羟基形成的分子间氢键进一步增加了葛根素的分子间作用力和熔点。这些化学结构特征使得葛根素的水溶性和脂溶性均较差。葛根素在水中的溶解度仅有0.462mg/ml,在磷酸盐缓冲液(ph 7.4)中的溶解度为7.56mg/ml。从20世纪50年代后期葛根素从野葛根中被分离出来后,其药理学特性己被广泛研究。它已广泛应用于治疗心脑血管疾病、阿兹海默症、帕金森、糖尿病、子宫内膜异位症、女性更年期综合征等等。
3.药代动力学实验结果表明,以50mg/kg葛根素溶液剂量灌胃后,在大鼠中的主要药代动力学参数为:最大血药浓度(cmax)为3.54
±
2.03mg/ml,药物浓度-时间曲线下面积(auc)为9.17
±
4.87mg*h*l-1,达峰时间(tmax)为0.68
±
0.37h,半衰期为1.70
±
0.60h。根据药代动力学分析,静脉注射后葛根素可以分布到体内的各个组织中,包括海马、心脏、肺、胃、肝、乳腺、肾、脾、胫骨、股骨、大脑皮层和纹状体。因此,葛根素在体内具有吸收快,能广泛分布到各个组织,但消除快,不易蓄积的特点。为了增加其生物利用度,研究者们从葛根素单体的角度出发,对结构和药物传输系统上加以改进。许庆兵等人利用化学法在4'位酚羟基引入氨基酸和葡萄糖苷基,合成了葛根素氨基酸类衍生物和7-羟基-4
′‑
(β-d-吡喃葡萄糖苷基)葛根素,大大提高了葛根素的水溶性。郝燕利用羟乙基化、酯化反应,在4'位酚羟基上合成琥珀酸单酯,并通过衍生物琥珀酸单酯上的羧基成盐,所得钠盐的水溶性较葛根素有极大提高。袁金伟利用磷酞化反应和乙基化反应,合成了7,4
′‑
二羟基葛根素,证明了葛根素7位羟基的反应活性高于4'位羟基。陈庆德等对葛根素葡糖糖基6
″
进行结构修饰,合成出了水溶性较好的葛根素磺酸钠(陈庆德,魏俊发,石先莹,et al.葛根素磺酸盐的合成[c]//葛根素磺酸盐的合成.中国化学会第二十五届学术年会.)。研究表明,对黄酮糖苷类化合物进行酰化或者酯化修饰,提高其脂溶性,有助于增强其肠渗透性和吸收。张首国等人在4
′
位羟基引入烷胺基烷基,合成了12个新的葛根素衍生物,提高了脂溶性,并发现其抗氧化性优于原化合物。但是,上述合成方法存在对环境不友好、纯化困难的问题,且合成的化合物在肠吸收阶段和体内生物利用度很少有人研究。因此,亟待开发一种绿色合成新型葛根素酸酯衍生物的新方法及其对合成的衍生物的生物活性进行评估。
技术实现要素:
[0004]
为了克服现有技术存在的不足,本发明的目的是提出绿色合成新型葛根素酸酯衍生物的方法及其应用。
[0005]
本发明筛选出具有催化效率的绿色溶剂,根据转化率和反应初速度优化出一种绿
色合成新型葛根素酸酯衍生物的方法,且所述的葛根素酸酯衍生物具有较好的生物利用度。本发明所述方法操作简单,环境友好,符合绿色化学理念;可解决目前葛根素脂溶性较差和生物利用度较差等问题,且所述的葛根素酸酯衍生物可用于制备抗心脑血管疾病药物。
[0006]
本发明的目的至少通过如下技术方案之一实现。
[0007]
一种葛根素酸酯衍生物,结构式如下:
[0008][0009]
上述的葛根素酸酯衍生物的制备方法,包括如下步骤:
[0010]
将葛根素和乙烯酯溶解于离子液体中,随后加入脂肪酶启动反应,置于恒温气浴振荡摇床中振荡反应;反应结束后,将反应混合物离心以除去脂肪酶,减压蒸馏除去溶剂,再经薄层层析分离得到所述葛根素酸酯衍生物。
[0011]
优选的,所述离子液体为1-乙基-3-甲基咪唑四氟硼酸盐,1-乙基-3-甲基咪唑醋酸盐,1-己基-3-甲基咪唑四氟硼酸盐,1-辛基-3-甲基咪唑四氟硼酸盐,1-丁基-3-甲基咪唑硫酸氢盐和1-丁基-3-甲基咪唑四氟硼酸盐中的一种以上。
[0012]
进一步优选的,所述离子液体为1-乙基-3-甲基咪唑醋酸盐,1-辛基-3-甲基咪唑四氟硼酸盐。
[0013]
优选的,所述脂肪酶为novozym 435,来源于c.antarctica的脂肪酶b固定在大孔
丙烯酸树脂上制备而成;lipozyme
im
rm,来源于r.miehei的脂肪酶固定在大孔阴离子交换树脂上制备而成;novozymes lipase tl100l;lipozyme
im
tl,来源于t.lanuginosus的脂肪酶通过物理吸附于硅胶颗粒中制备而成;amano ps-im,来源于p.cepacia固定在硅藻土上制备而成;猪胰酶中的一种以上。
[0014]
进一步优选的,所述所述脂肪酶为novozym 435、novozymes lipase tl100l。
[0015]
优选地,所述乙烯酯为碳的个数为4-16的乙烯酯,所述乙烯酯作为酰基供体。
[0016]
进一步优选的,所述乙烯酯为醋酸乙烯酯,丙酸乙烯酯,丁酸乙烯酯,己酸乙烯酯,辛酸乙烯酯,月桂酸乙烯酯,肉豆蔻酸乙烯酯。
[0017]
优选的,所述葛根素与乙烯酯的摩尔比为1:1-1:50,脂肪酶量与离子液体的质量体积比例为1:1-1:40mg/ml。
[0018]
进一步优选的,所述葛根素与乙烯酯的摩尔比为1:30,脂肪酶量与离子液体的质量体积比例为1:2mg/ml。
[0019]
优选的,所述反应温度为30-80℃,反应时间为0.5-24h。
[0020]
优选的,所述离心的转速为3000~18000rpm,离心的时间为5~40min。
[0021]
进一步优选的,所述离心处理的转速为12000rpm,离心处理的时间为10min。
[0022]
优选的,所述葛根素乙酸酯、丙酸酯、丁酸酯、己酸酯和辛酸酯的薄层层析的层析液按体积组成为:氯仿/甲醇/水=8:2:0.3;葛根素月桂酸酯的层析液按体积组成为:乙酸乙酯/丙酮=10:1;葛根素肉豆蔻酸酯的层析液按体积组成为:乙酸乙酯/丙酮=9:1。
[0023]
上述的葛根素酸酯衍生物在制备抗心脑血管疾病药物中的应用。
[0024]
通过以下方法对上述制备的葛根素酸酯衍生物进行测试:
[0025]
选取健康的雄性sprague-dawley(sd)大鼠,spf级,体重为180-220g,用含有0.5%羧甲基纤维素钠的生理盐水溶解所述的葛根素酸酯衍生物。在灌胃前后每组随机抽取动物进行眼眶后静脉窦取血,离心,取上清血浆,保存于-80℃冰箱待测。另于不同时间点每组随机抽取动物用戊巴比妥钠45mg/kg腹腔注射麻醉,取心、肝、脾、肺、肾、大脑皮层、海马及纹状体样本。建立检测方法,包括:色谱专属性检测、建立标准曲线和线性范围、测定精密度和准确度。根据hplc检测药物峰面积代入标准曲线,计算各个样本中葛根素及其酸酯衍生物的含量,在根据药物浓度-时间数据,计算出药代动力学参数。
[0026]
优选的,将所有的大鼠随机分别1个对照组和8个实验组,每组20只,每只大鼠灌胃葛根素及其酸酯衍生物,每组随机抽取动物进行眼眶后静脉窦取血0.5ml,并置于抗凝管中。静置半小时后,3500rpm 4℃条件下离心10min,取上清血浆,保存于-80℃冰箱备用。
[0027]
优选的,取200μl血浆与30μl 3mol/l甲酸溶液混合,随后加入1.6ml乙酸乙酯和甲醇的混合液(3:1,v/v)混合均匀,涡旋10min,在12,000r/min、4℃下离心10min,取上清液用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。
[0028]
优选的,将各个组织解冻后,迅速称量组织样本。用生理盐水冲洗组织,并用滤纸擦开。将各个组织样本(0.2g)与1ml生理盐水混合、匀浆,随后加入乙酸乙酯和甲醇的混合液重复萃取两次,收集萃取液并用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。
[0029]
优选的,hplc使用agilent 1290系统,在c18柱(4.6mm
×
250mm,5μm)上进行分离,利用紫外检测器在254nm下进行检测。色谱条件:(1)流动相组成:甲醇和水;(2)进样量:20μ
l;(3)流速:0.5ml/min;(4)柱温:30℃;(5)葛根素(pu),葛根素醋酸酯(pa),葛根素丙酸酯(pp),葛根素丁酸酯(pb),葛根素己酸酯(ph),葛根素辛酸酯(po),葛根素月桂酸酯(pl)和葛根素肉豆蔻酸酯(pm)的流动相比例为甲醇/水(35/65,35/65,55/55,55/55,65/35,80/20,80/20,90/10)。
[0030]
优选地,取500μl空白血浆和组织萃取液,添加了500μl浓度为1mg/ml、0.2mg/ml、0.04mg/ml、0.008mg/ml、0.0016mg/ml、0.00032mg/ml的葛根素及其酸酯衍生物的标准工作液,利用hplc所得到的峰面积结果,以葛根素峰面积为纵坐标,葛根素对照品浓度(mg/ml)为横坐标制备标准曲线和相关系数。
[0031]
与现有技术相比,本发明具有如下优点和有益效果:
[0032]
(1)本发明提供的绿色合成新型葛根素酸酯衍生物的方法及其应用的方法,利用绿色溶剂和脂肪酶催化葛根素酰化,制备出有着更好的脂溶性和生物利用度的酸酯衍生物,该方法中底物转化率可达90%以上。
[0033]
(2)与化学法和其他酶法相比,本发明提供的绿色合成新型葛根素酸酯衍生物的方法及其应用的方法避免了大量有机试剂的使用,对环境友好,符合绿色化学理念,且具有底物转化率高,反应选择性好,条件温和,可重复利用等优点。
[0034]
(3)本发明填补了国内外关于葛根素酸酯衍生物药代动力学研究的空白,为今后在应用于抗心脑血管疾病上提供了理论基础。
附图说明
[0035]
图1为实施例1得到的葛根素丙酸酯结构式;
[0036]
图2为实施例1中葛根素丙酸酯的质谱分析图;
[0037]
图3为实施例中大鼠中血液样本的色谱图;空白血液色谱图(a);空白血液中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.539min(b);sd大鼠口服葛根素及其酸酯衍生物后血液样本色谱图,葛根素的保留时间为puerarin=9.924min(c);
[0038]
图4为实施例中大鼠中海马体样本的色谱图;空白海马体色谱图(a);空白海马体组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.676min(b);sd大鼠口服葛根素及其酸酯衍生物后海马体样本色谱图,葛根素的保留时间为puerarin=10.197min(c);
[0039]
图5为实施例中大鼠中肝脏组织样本的色谱图;空白肝组织色谱图(a);空白肝组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.796min(b);sd大鼠口服葛根素及其酸酯衍生物后肝组织样本色谱图,葛根素的保留时间为puerarin=10.001min(c);
[0040]
图6为实施例中大鼠中心脏组织样本的色谱图;空白心脏组织色谱图(a);空白心脏组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.034min(b);sd大鼠口服葛根素及其酸酯衍生物后心脏组织样本色谱图,葛根素的保留时间为puerarin=9.743min(c);
[0041]
图7为实施例中大鼠中纹状体组织样本的色谱图;空白纹状体色谱图(a);空白纹状体组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.729min(b);sd大鼠口服葛根素及其酸酯衍生物后纹状体样本色谱图,葛根素的保留时间为puerarin=
9.805min(c);
[0042]
图8为实施例中大鼠中大脑皮层组织样本的色谱图;空白大脑皮层色谱图(a);空白大脑皮层组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.643min(b);sd大鼠口服葛根素及其酸酯衍生物后大脑皮层样本色谱图,葛根素的保留时间为puerarin=9.988min(c);
[0043]
图9为实施例中大鼠中肾脏组织样本的色谱图;空白肾组织色谱图(a);空白肾组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为10.192min(b);sd大鼠口服葛根素及其酸酯衍生物后肾组织样本色谱图,葛根素的保留时间为puerarin=10.124min(c);
[0044]
图10为实施例中大鼠中肺组织样本的色谱图;空白肺组织色谱图(a);空白肺组织中加入葛根素标品(0.5mg/ml)的色谱图,葛根素的保留时间为9.072min(b);sd大鼠口服葛根素及其酸酯衍生物后肺组织样本色谱图,葛根素的保留时间为puerarin=9.796min(c);
[0045]
图11为实施例中大鼠中脾脏组织样本的色谱图;空白脾组织色谱图(a);空白脾组织中加入葛根素标品(0.0008mg/ml)的色谱图,葛根素的保留时间为10.061min(b);sd大鼠口服葛根素及其酸酯衍生物后脾组织样本色谱图,葛根素的保留时间为puerarin=10.238min(c);
[0046]
图12为实施例2得到的葛根素己酸酯结构式;
[0047]
图13为实施例2中葛根素己酸酯的质谱分析图;
[0048]
图14为实施例3得到的葛根素肉豆蔻酸酯结构式;
[0049]
图15为实施例3中葛根素肉豆蔻酸酯的质谱分析图;
[0050]
图16为葛根素和实施例1-3口服葛根素及其酸酯衍生物的大鼠血液药时曲线图。
具体实施方式
[0051]
以下结合实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器未注明生产厂商者,视为可以通过市售购买得到的常规产品。
[0052]
以下实施例中所采用的检测方法为hplc测定方法。
[0053]
所述hplc测定方法采用的色谱柱:agilent c18(250mm
×
4.6mm);
[0054]
流动相:葛根素(pu),葛根素醋酸酯(pa),葛根素丙酸酯(pp),葛根素丁酸酯(pb),葛根素己酸酯(ph),葛根素辛酸酯(po),葛根素月桂酸酯(pl)和葛根素肉豆蔻酸酯(pm)的流动相比例为甲醇/水(35/65,35/65,55/55,55/55,65/35,80/20,80/20,90/10),流动相的流速为0.5ml/min;
[0055]
检测波长:254nm,进样量20μl,柱温30℃的条件下,进行等度洗脱。
[0056]
实施例1
[0057]
绿色合成新型葛根素酸酯衍生物(pp)的方法及其应用,包括如下步骤:
[0058]
(1)将20mmol/l葛根素和600mmol/l丙酸乙烯酯完全溶解于1ml的1-乙基-3-甲基咪唑醋酸盐离子液体中。随后加入2mg/ml novozym 435脂肪酶启动反应。经反应瓶置于恒温气浴振荡摇床(200r/min,50℃)中振荡反应24h。利用薄层层析柱进行分离纯化。将反应完成的反应混合物通过离心(12,000r/min,10min)和真空减压蒸馏除去脂肪酶催化剂,获
得粗产物。随后,将收集含产物的层析液(氯仿/甲醇/水=8:2:0.3(v/v))经减压蒸馏和甲醇结晶获得淡黄色或黄色粉末状物质。其葛根素丙酯的产率为90%,纯度为95%。
[0059]
(2)设置2组实验大鼠(健康的雄性sprague-dawley(sd)大鼠,spf级,体重为180-220g),一组对照组,每组20只。葛根素组、葛根素丙酸酯组的灌胃剂量为40mg/kg、47.53mg/kg。每组用含有0.5%羧甲基纤维素钠的生理盐水溶解。在灌胃前后每组随机抽取动物进行眼眶后静脉窦取血0.5ml,并置于抗凝管中。静置半小时后,3500rpm 4℃条件下离心10min,取上清血浆,保存于-80℃冰箱待测。另于不同时间点每组随机抽取动物用戊巴比妥钠45mg/kg腹腔注射麻醉,取心、肝、脾、肺、肾、大脑皮层、海马及纹状体,保存于﹣80℃冰箱待测。
[0060]
(3)取200μl血浆与30μl 3mol/l甲酸溶液混合,随后加入1.6ml乙酸乙酯和甲醇的混合液(3:1,v/v)混合均匀,涡旋10min,在12,000r/min、4℃下离心10min,取上清液用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。将各个组织解冻后,迅速称量组织样本。用生理盐水冲洗组织,并用滤纸擦开。将各个组织样本(0.2g)与1ml生理盐水混合、匀浆,随后加入乙酸乙酯和甲醇的混合液重复萃取两次,收集萃取液并用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。
[0061]
(4)取500μl空白血浆和组织萃取液,添加了500μl浓度为1mg/ml、0.2mg/ml、0.04mg/ml、0.008mg/ml、0.0016mg/ml、0.00032mg/ml的葛根素及其葛根素丙酸酯的标准工作液,利用hplc所得到的峰面积结果,以葛根素峰面积为纵坐标,葛根素对照品浓度(mg/ml)为横坐标制备标准曲线和相关系数。将上述样本在同一色谱条件下同一天连续进样多次,分别计算葛根素的日内准确度和精确度;同样地,将上述样本再同一色谱条件下连续三天进样,分别葛根素的日间准确度和精确度。根据hplc检测药物峰面积代入标准曲线,计算各个样本中葛根素及其酸酯衍生物的含量,在根据药物浓度-时间数据,计算出药代动力学参数。
[0062]
效果验证
[0063]
在酰化反应和分离纯化后得到了葛根素丙酸酯,其结构如图1所示,其质谱分析图如图2所示,信号强度最强的离子峰的m/z为481.1115,而葛根素的分子质量为416,故该峰应该是pa:c
23h22
nao
10
,481.11m/z[m na]
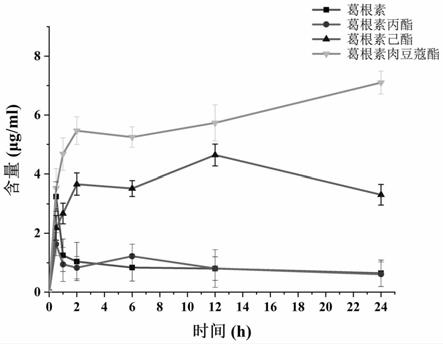
分子离子加钠的结果。本研究中方法学验证结果表明,所建立的方法具有良好的线性、精密度和准确度和选择性,符合葛根素酸酯衍生物的药代动力学研究的要求,如图3-11。本研究中的hplc所测的标准曲线和准确度、精确度如表1和表2所示。口服葛根素丙酸酯后,与葛根素本身相比,显著地提高葛根素在血液(图16)和各个组织中的auc值,极大地增加了葛根素的吸收,其在大脑皮层、海马体和纹状体的auc值增加了5.48、11.75和6.73倍(表3、表4)。
[0064]
表1葛根素在各个组织中的标准曲线
[0065][0066]
表2葛根素测定方法的精密度
[0067][0068][0069]
表3饲喂葛根素及其酸酯衍生物后大鼠组织中的药代动力学参数
[0070][0071]
[0072]
表4饲喂葛根素及其酸酯衍生物后大鼠大脑中的药代动力学参数
[0073][0074][0075]
实施例2
[0076]
绿色合成新型葛根素酸酯衍生物(ph)的方法及其应用,包括如下步骤:
[0077]
(1)将20mmol/l葛根素和800mmol/l己酸乙烯酯完全溶解于1ml的1-乙基-3-甲基咪唑醋酸盐([emim][oac])离子液体中。随后加入2mg/ml novozym435脂肪酶启动反应。经反应瓶置于恒温气浴振荡摇床(200r/min,50℃)中振荡反应24h。利用薄层层析柱进行分离纯化。将反应完成的反应混合物通过离心(12,000r/min,10min)和真空减压蒸馏除去脂肪酶催化剂,获得粗产物。随后,将收集含产物的层析液(氯仿/甲醇/水=8:2:0.3(v/v))经减压蒸馏和甲醇结晶获得淡黄色或黄色粉末状物质。葛根素己酯的产率为88%,纯度为94%。
[0078]
(2)设置2组实验大鼠(健康的雄性sprague-dawley(sd)大鼠,spf级,体重为180-220g),一组对照组,每组20只。葛根素组、葛根素己酸酯组、的灌胃剂量为40mg/kg、51.56mg/kg。每组用含有0.5%羧甲基纤维素钠的生理盐水溶解。在灌胃前后每组随机抽取动物进行眼眶后静脉窦取血0.5ml,并置于抗凝管中。静置半小时后,3500rpm 4℃条件下离
心10min,取上清血浆,保存于-80℃冰箱待测。另于不同时间点每组随机抽取动物用戊巴比妥钠45mg/kg腹腔注射麻醉,取心、肝、脾、肺、肾、大脑皮层、海马及纹状体,保存于﹣80℃冰箱待测。
[0079]
(3)取200μl血浆与30μl 3mol/l甲酸溶液混合,随后加入1.6ml乙酸乙酯和甲醇的混合液(3:1,v/v)混合均匀,涡旋10min,在12,000r/min、4℃下离心10min,取上清液用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。将各个组织解冻后,迅速称量组织样本。用生理盐水冲洗组织,并用滤纸擦开。将各个组织样本(0.2g)与1ml生理盐水混合、匀浆,随后加入乙酸乙酯和甲醇的混合液重复萃取两次,收集萃取液并用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。
[0080]
(4)取500μl空白血浆和组织萃取液,添加了500μl浓度为1mg/ml、0.2mg/ml、0.04mg/ml、0.008mg/ml、0.0016mg/ml、0.00032mg/ml的葛根素及其葛根素己酸酯的标准工作液,利用hplc所得到的峰面积结果,以葛根素峰面积为纵坐标,葛根素对照品浓度(mg/ml)为横坐标制备标准曲线和相关系数。将上述样本在同一色谱条件下同一天连续进样多次,分别计算葛根素的日内准确度和精确度;同样地,将上述样本再同一色谱条件下连续三天进样,分别葛根素的日间准确度和精确度。根据hplc检测药物峰面积代入标准曲线,计算各个样本中葛根素及其酸酯衍生物的含量,在根据药物浓度-时间数据,计算出药代动力学参数。
[0081]
效果验证
[0082]
在酰化反应和分离纯化后得到了葛根素己酸酯,结构如图12所示,其质谱分析图如图13所示,信号强度最强的离子峰应该是ph:c
27h30o10
,515.17m/z[m h]
。本研究中方法学验证结果表明,所建立的方法具有良好的线性、精密度和准确度和选择性,符合葛根素酸酯衍生物的药代动力学研究的要求,如图3-11。本研究中的hplc所测的标准曲线和准确度、精确度如表1和表2所示。口服葛根素己酸酯后,与葛根素本身相比,显著地提高葛根素在血液(图16)和各个组织中的auc值,极大地增加了葛根素的吸收,其在大脑皮层、海马体和纹状体的auc值增加了11.90、13.70和27.27倍(表3、表4)。
[0083]
实施例3
[0084]
绿色合成新型葛根素酸酯衍生物的方法(pm)及其应用,包括如下步骤:
[0085]
(1)将20mmol/l葛根素和600mmol/l肉豆蔻酸乙烯酯完全溶解于1ml的1-乙基-3-甲基咪唑醋酸盐([emim][oac])离子液体中。随后加入2mg/ml novozym 435脂肪酶启动反应。经反应瓶置于恒温气浴振荡摇床(200r/min,50℃)中振荡反应24h。利用薄层层析柱进行分离纯化。将反应完成的反应混合物通过离心(12,000r/min,10min)和真空减压蒸馏除去脂肪酶催化剂,获得粗产物。随后,将收集含产物的层析液(乙酸乙酯/丙酮=9:1(v/v))经减压蒸馏和甲醇结晶获得淡黄色或黄色粉末状物质。葛根素肉豆蔻酯的产率为85%,纯度为93%。
[0086]
(2)设置2组实验大鼠(健康的雄性sprague-dawley(sd)大鼠,spf级,体重为180-220g),一组对照组,每组20只。葛根素组、葛根素肉豆蔻酸酯组的灌胃剂量为40mg/kg、62.33mg/kg。每组用含有0.5%羧甲基纤维素钠的生理盐水溶解。在灌胃前后每组随机抽取动物进行眼眶后静脉窦取血0.5ml,并置于抗凝管中。静置半小时后,3500rpm 4℃条件下离心10min,取上清血浆,保存于-80℃冰箱待测。另于不同时间点每组随机抽取动物用戊巴比
妥钠45mg/kg腹腔注射麻醉,取心、肝、脾、肺、肾、大脑皮层、海马及纹状体,保存于﹣80℃冰箱待测。
[0087]
(3)取200μl血浆与30μl 3mol/l甲酸溶液混合,随后加入1.6ml乙酸乙酯和甲醇的混合液(3:1,v/v)混合均匀,涡旋10min,在12,000r/min、4℃下离心10min,取上清液用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。将各个组织解冻后,迅速称量组织样本。用生理盐水冲洗组织,并用滤纸擦开。将各个组织样本(0.2g)与1ml生理盐水混合、匀浆,随后加入乙酸乙酯和甲醇的混合液重复萃取两次,收集萃取液并用氮气吹干。最后加入100μl甲醇复溶并离心,取上清液进行hplc分析。
[0088]
(4)取500μl空白血浆和组织萃取液,添加了500μl浓度为1mg/ml、0.2mg/ml、0.04mg/ml、0.008mg/ml、0.0016mg/ml、0.00032mg/ml的葛根素及其葛根素肉豆蔻酸酯的标准工作液,利用hplc所得到的峰面积结果,以葛根素峰面积为纵坐标,葛根素对照品浓度(mg/ml)为横坐标制备标准曲线和相关系数。将上述样本在同一色谱条件下同一天连续进样多次,分别计算葛根素的日内准确度和精确度;同样地,将上述样本再同一色谱条件下连续三天进样,分别葛根素的日间准确度和精确度。根据hplc检测药物峰面积代入标准曲线,计算各个样本中葛根素及其酸酯衍生物的含量,在根据药物浓度-时间数据,计算出药代动力学参数。
[0089]
效果验证
[0090]
在酰化反应和分离纯化后得到了葛根素肉豆蔻酸酯,结构如图14所示,其质谱分析图如图15所示,信号强度最强的离子峰应该是pm:c
35h46o10
,627.29m/z[m h]
。本研究中方法学验证结果表明,所建立的方法具有良好的线性、精密度和准确度和选择性,符合葛根素酸酯衍生物的药代动力学研究的要求,如图3-11。本研究中的hplc所测的标准曲线和准确度、精确度如表1和表2所示。口服葛根素肉豆蔻酸酯后,与葛根素本身相比,显著地提高葛根素在血液(图16)和各个组织中的auc值,极大地增加了葛根素的吸收,其在大脑皮层、海马体和纹状体的auc值增加了48.51、42.36和89.48倍(表3、表4)。
[0091]
以上实施例仅为本发明较优的实施方式,仅用于解释本发明,而非限制本发明,本领域技术人员在未脱离本发明精神实质下所作的改变、替换、修饰等均应属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。