1.本发明属于药物化学和药物治疗学领域,涉及一种雷公藤红素衍生物及其制备方法与在 制备抗癌药物中的应用。
技术背景
2.癌症是影响人类健康与寿命的主要疾病,现已成为全球重要的公共卫生问题之一。国际 癌症研究报告数据显示,预估2020年全球新发癌症病例1929万例,因癌症死亡人数达995 万。据估计,2020年中国癌症新发病例约456万例,死亡病例300万例。癌症是导致人群 期望寿命损失的首要疾病因素,致使越来越多的人关注癌症的治疗。
3.最常见的癌症治疗方法主要有手术、化疗、放疗及生物疗法,其中化疗应用最广泛。临 床上用于治疗癌症的药物种类繁多,例如生物烷化剂类、抗代谢类、抗生素类、天然产物 等。目前,已有多种天然活性成分被证实具有抗肿瘤活性,但已上市的活性良好的天然抗癌 药物较少。因此,开发活性优异的天然产物已经成为癌症治疗的迫切需求。
4.雷公藤红素(celastrol,cel),又名南蛇藤素,是从中药雷公藤根皮中分离得到的五环 三萜类化合物。大量研究发现雷公藤红素具有多种生物活性,如抗炎、抗肿瘤、抗氧化、抗 真菌、抗神经退行性疾病等。其中,雷公藤红素的抗肿瘤活性已成为近年来研究的热点。体 内外活性研究表明,雷公藤红素可以有效抑制多种肿瘤的生长,如多发性骨髓瘤、黑色素 瘤、肝癌、胃癌、前列腺癌、肾癌、非小细胞肺癌、胶质瘤和乳腺癌等,是一种广谱的抗肿 瘤活性化合物。研究表明,雷公藤红素可以调节多种靶点,发挥其抗肿瘤作用。
[0005][0006]
尽管雷公藤红素具有较明确的抗肿瘤活性,但其活性及成药性均有待提高,也一直制约 着其发展。因此,以其为先导物进行结构修饰改造,寻找活性更强、成药性更佳、具有靶向 性的衍生物非常必要。
技术实现要素:
[0007]
尽管cel具有不错的抗肿瘤活性,但其无明确的靶向性。肿瘤细胞线粒体膜电位高于 正常细胞,其内膜电位约为-180mv,对亲脂性的阳离子具有很强的吸引力。因此,本发明 的目的在于将三苯基膦或三环己基膦引入cel得到一类连接离域亲脂阳离子的雷公藤红素 衍生物,通过亲脂性阳离子三苯基膦和三环己基膦,使雷公藤红素衍生物通过静电作用被肿 瘤细胞线粒体吸引,在线粒体基质中积聚,引起肿瘤细胞凋亡。
[0008]
本发明的目的是通过以下技术方案实现的:
[0009]
一种如式ⅳ所示的雷公藤红素衍生物:
[0010][0011]
其中,l选自饱和烷烃链-(ch2)
n-、不饱和芳香烃片段n为 2~7的整数;
[0012]
r选自
[0013]
优选的,n为3~5的整数。
[0014]
作为本发明所述的雷公藤红素衍生物的其中一个优选技术方案,所述的雷公藤红素衍生 物为如式ⅰ所示的雷公藤红素三苯基膦衍生物:
[0015][0016]
其中,l选自饱和烷烃链-(ch2)
n-、不饱和芳香烃片段
[0017]
n为2~7的整数,优选为3~5的整数。
[0018]
作为本发明所述的雷公藤红素衍生物的其中一个优选技术方案,所述的雷公藤红素衍生 物为如式ⅱ所示的雷公藤红素三环己基膦衍生物:
[0019]
[0020]
其中,l选自饱和烷烃链-(ch2)
n-、不饱和芳香烃片段
[0021]
n为2~7的整数,优选为3~5的整数。
[0022]
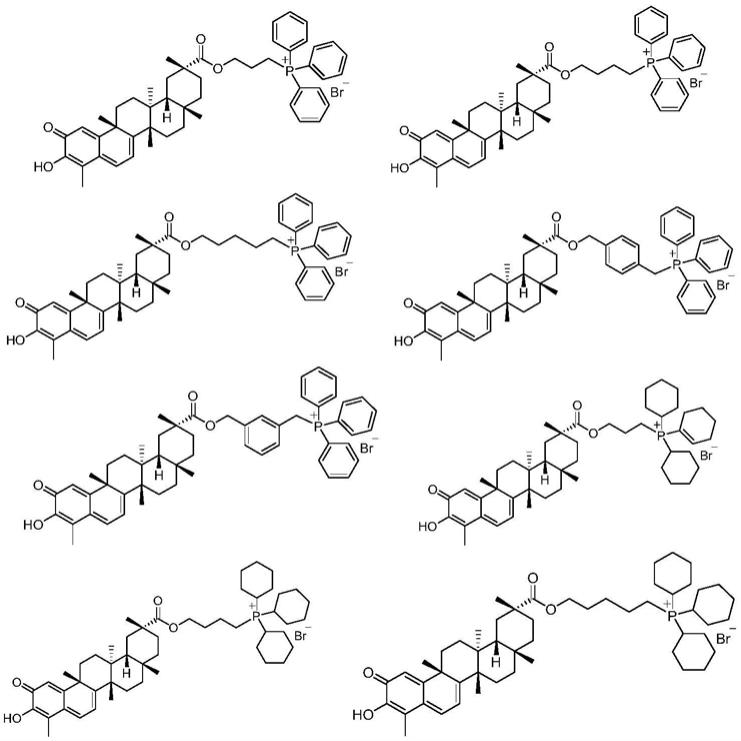
具体地讲,本发明所述的雷公藤红素衍生物选自以下化合物:
[0023][0024]
本发明的另一目的在于提供所述的雷公藤红素衍生物的制备方法,合成路线如下:
[0025][0026]
包括:式ⅲ所示的中间体与三苯基膦或三环己基膦反应得到雷公藤红素衍生物;
其中, 式ⅲ所示的中间体与三苯基膦或三环己基膦摩尔比为1:1.2~1:1.5;反应溶剂为乙腈或n,n
‑ꢀ
二甲基甲酰胺(dmf)或二甲基亚砜(dmso);反应温度为60~70℃。
[0027]
作为本发明cel衍生物的制备方法的进一步技术方案,反应结束后,往反应液加入与 反应溶剂等体积的水稀释,用二氯甲烷萃取3次,合并有机层,饱和食盐水溶液洗涤,无水 硫酸钠干燥,过滤,浓缩有机相,以二氯甲烷:甲醇=50:1v/v为洗脱剂,硅胶柱层析得到雷 公藤红素衍生物。
[0028]
药理实验表明,本发明雷公藤红素衍生物对肿瘤细胞的增殖抑制作用显著优于雷公藤红 素。本发明雷公藤红素衍生物可以单独或与临床上常用的抗肿瘤药物如抗代谢药物、烷化 剂、抗肿瘤抗生素、抗肿瘤植物药、激素类抗肿瘤药联合应用,另外还可以与放射治疗联合 应用。因此,本发明的另一个目的在于提供所述的雷公藤红素衍生物在制备抗肿瘤药物中的 用途。
[0029]
所述的肿瘤为非小细胞肺癌、结肠癌和乳腺癌。
[0030]
本发明的另一个目的是提供一种药物组合物,含有本发明有效剂量的雷公藤红素衍生 物,辅以药学上可接受的载体,制成任何药学上可接受的剂型,,以供临床口服、注射或局 部用药。
[0031]
所述的剂型选自片剂、胶囊剂、滴丸、颗粒剂、粉剂、锭剂、水性或油性悬浮剂、注射 剂、贴剂、纳米制剂。
[0032]
优选的,所述的药物组合物中还含有抗代谢药物、烷化剂、抗肿瘤抗生素、抗肿瘤植物 药、激素类抗肿瘤药。
[0033]
本发明的有益效果:
[0034]
本发明雷公藤红素衍生物的制备方法反应条件温和,所用试剂低毒,原料易得,后处理 方便,产率较高。
[0035]
药理实验表明,本发明雷公藤红素衍生物通过静电作用被肿瘤细胞线粒体吸引,在线粒 体基质中积聚,引起肿瘤细胞凋亡,其抗肿瘤活性显著优于雷公藤红素,可作为抗肿瘤药物 应用。
具体实施方式
[0036]
下面列举一系列实施例进一步阐明本发明的技术方案。这些实施例是例证性的,不应当 理解为对本发明的限制。
[0037]
雷公藤红素衍生物有两条合成路线。
[0038]
路线a:式ⅲ所示的中间体与三苯基膦反应得到结构如式ⅰ所示的雷公藤红素三苯基膦 衍生物。
[0039]
[0040]
路线b:式ⅲ所示的中间体与三环己基膦反应得到结构如式ⅱ所示的雷公藤红素三环己 基膦衍生物。
[0041][0042]
式ⅲ所示的中间体的合成路线如下:
[0043][0044]
实施例1:cel溴代丙烷中间体的制备
[0045][0046]
135mg(0.3mmol)cel溶于15ml n,n-二甲基甲酰胺中,加入1,3-二溴丙烷121mg(0.6 mmol)、nahco
3 125mg(1.2mmol),60℃搅拌反应8h,加入15ml水稀释,用二氯甲烷萃取 三次(每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓 缩有机相,硅胶(200-300目)柱层析(洗脱剂为石油醚:乙酸乙酯=20:1v/v),真空干燥过夜,得 到cel溴代丙烷中间体(橘红色粉末,121mg,产率70.1%)。
[0047]1h nmr(300mhz,dmso,tms),δ7.06(1h,d,j=7.14hz),6.41(1h,s),6.37(1h,d,j =7.12hz),4.06(2h,m),3.64(2h,m),2.21(3h,s),1.43(3h,s),1.27(3h,s),1.12(3h,s),0.96 (3h,s),0.42(3h,s).
13
c nmr(75mhz,dmso,tms),δ178.21,164.66,135.14,133.72, 130.63,118.21,117.34,111.97,77.48,76.63,45.00,42.91,40.50,39.39,38.26,36.27,34.79,33.50, 33.08,31.56,29.66,21.62,18.69,10.32.esi-ms[m-br]
:571.6.
[0048]
实施例2:化合物i-1的制备
[0049][0050]
114mg(0.2mmol)cel溴代丙烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三苯基膦 82mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物i
‑ꢀ
1(橘红色粉末,87mg,产率51.7%)。
[0051]1h nmr(300mhz,dmso,tms),δ7.80(15h,m),7.06(1h,d,j=7.14hz),6.97(1h,s), 6.41(1h,s),6.37(1h,d,j=7.12hz),4.26(2h,m),3.94(2h,m),2.22(3h,s),1.42(3h,s), 1.26(3h,s),1.11(3h,s),0.96(3h,s),0.42(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.21,164.66,135.14,133.72,133.58,130.63,130.46,118.21,117.34,111.97,77.48,77.05, 76.63,45.00,44.11,42.91,40.50,39.39,38.26,36.27,34.79,33.50,33.08,31.56,30.48,29.66, 21.62,18.69,10.32.esi-ms[m-br]
:753.4.
[0052]
实施例3:cel溴代丁烷中间体的制备
[0053][0054]
135mg(0.3mmol)cel溶于15ml n,n-二甲基甲酰胺中,加入1,4-二溴丁烷130mg(0.6 mmol)、nahco
3 125mg(1.2mmol),60℃搅拌反应8h,加入15ml水稀释,用二氯甲烷萃取 三次(每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓 缩有机相,硅胶(200-300目)柱层析(洗脱剂为石油醚:乙酸乙酯=20:1v/v),真空干燥过夜,得 到cel溴代丁烷中间体(橘红色粉末,128mg,产率72.7%)。
[0055]1h nmr(300mhz,dmso,tms),δ7.06(1h,d,j=7.14hz),6.41(1h,s),6.37(1h,d,j =7.16hz),4.09(2h,m),3.66(2h,m),2.23(3h,s),1.45(3h,s),1.28(3h,s),1.10(3h,s),0.96 (3h,s),0.43(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.21,164.66,135.14,133.58, 130.46,118.21,117.34,111.97,77.48,76.63,45.00,44.11,40.50,39.33,38.45,36.29,34.79,33.08, 31.56,30.69,29.55,21.37,18.81,10.37.esi-ms[m-br]
:585.6.
[0056]
实施例4:化合物i-2的制备
[0057][0058]
117mg(0.2mmol)cel溴代丁烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三苯基膦 82mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物i
‑ꢀ
2(橘红色粉末,90mg,产率53.2%)。
[0059]1h nmr(300mhz,dmso,tms)δ7.81(15h,m),7.07(1h,d,j=7.14hz),6.98(1h,s), 6.43(1h,s),6.37(1h,d,j=7.16hz),4.28(2h,m),3.95(2h,m),2.23(3h,s),1.43(3h,s), 1.27(3h,s),1.10(3h,s),0.94(3h,s),0.43(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.33,164.86,135.27,133.81,133.65,130.78,130.59,118.36,117.47,111.97,77.47,77.01, 76.68,45.02,44.26,42.97,40.62,39.44,38.21,36.27,34.85,33.49,33.05,31.63,30.45,29.85, 21.63,18.74,10.38.esi-ms[m-br]
:767.4.
[0060]
实施例5:cel溴代戊烷中间体的制备
[0061][0062]
135mg(0.3mmol)cel溶于15ml n,n-二甲基甲酰胺中,加入1,5-二溴戊烷138mg(0.6 mmol)、nahco
3 125mg(1.2mmol),60℃搅拌反应8h,加入15ml水稀释,用二氯甲烷萃 取三次(每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤, 浓缩有机相,硅胶(200-300目)柱层析(洗脱剂为石油醚:乙酸乙酯=20:1v/v),真空干燥过夜, 得到cel溴代戊烷中间体(橘红色粉末,130mg,产率72.2%)。
[0063]1h nmr(300mhz,dmso,tms),δ7.06(1h,d,j=7.16hz),6.42(1h,s),6.37(1h,d,j =7.12hz),4.07(2h,m),3.61(2h,m),2.21(3h,s),1.42(3h,s),1.25(3h,s),1.13(3h,s),0.98 (3h,s),0.41(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.36,164.85,135.42,133.68, 130.77,118.21,117.34,111.97,77.35,76.63,45.00,44.39,40.50,39.39,38.26,36.27,34.63,33.28, 31.56,30.78,29.61,21.32,18.69,10.48.esi-ms[m-br]
:599.6.
[0064]
实施例6:化合物i-3的制备
[0065][0066]
120mg(0.2mmol)cel溴代戊烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三苯基膦 82mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物i
‑ꢀ
3(橘红色粉末,95mg,产率55.3%)。
[0067]1h nmr(300mhz,dmso,tms)δ7.79(15h,m),7.06(1h,d,j=7.16hz),6.95(1h,s), 6.45(1h,s),6.36(1h,d,j=7.12hz),4.25(2h,m),3.93(2h,m),2.21(3h,s),1.43(3h,s), 1.28(3h,s),1.10(3h,s),0.97(3h,s),0.43(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.79,164.88,135.24,133.83,133.65,130.69,130.22,118.14,117.30,111.81,77.45,77.05, 76.75,45.25,44.17,42.82,40.35,39.48,38.35,36.56,34.87,33.45,33.03,31.23,30.76,29.56, 21.74,18.96,10.53.esi-ms[m-br]
:781.4.
[0068]
实施例7:cel溴代对苯芳香烃中间体的制备
[0069][0070]
135mg(0.3mmol)cel溶于15ml n,n-二甲基甲酰胺中,加入1,4-二(溴甲基)苯158 mg(0.6mmol)、nahco
3 125mg(1.2mmol),60℃搅拌反应8h,加入15ml水稀释,用二氯 甲烷萃取三次(每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥, 过滤,浓缩有机相,硅胶(200-300目)柱层析(洗脱剂为石油醚:乙酸乙酯=20:1v/v),真空干燥 过夜,得到cel溴代对苯芳香烃中间体(橘红色粉末,135mg,产率70.7%)。
[0071]1h nmr(300mhz,dmso,tms),δ7.34(2h,d,j=8.24hz),7.21(2h,d,j=8.22hz), 7.06(1h,d,j=7.14hz),6.97(1h,s),6.41(1h,s),6.37(1h,d,j=7.12hz),4.15(2h,m),3.74 (2h,m),2.24(3h,s),1.45(3h,s),1.28(3h,s),1.13(3h,s),0.95(3h,s),0.43(3h,s).
13
c nmr (75mhz,dmso,tms),δppm:δ178.47,164.92,135.53,133.61,130.82,118.42,117.39, 111.97,77.51,76.84,45.18,42.64,40.55,39.45,38.58,36.29,34.79,33.28,31.74,30.51,29.87, 21.74,18.89,10.51.esi-ms[m-br]
:633.6.
[0072]
实施例8:化合物i-4的制备
[0073][0074]
127mg(0.2mmol)cel溴代对苯芳香烃中间体溶于15ml n,n-二甲基甲酰胺中,加入三 苯基膦82mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每 次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机 相,硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物 i-4(橘红色粉末,98mg,产率54.7%)。
[0075]1h nmr(300mhz,dmso,tms)δ7.81(15h,m),7.34(2h,d,j=8.20hz),7.21(2h,d,j =8.22hz),7.06(1h,d,j=7.14hz),6.97(1h,s),6.41(1h,s),6.37(1h,d,j=7.12hz),4.26(2h, m),3.94(2h,m),2.22(3h,s),1.42(3h,s),1.26(3h,s),1.11(3h,s),0.96(3h,s),0.44(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.07,177.75,171.16,165.27,165.23,145.90, 136.06,135.05,134.49,131.89,130.26,128.80,127.04,119.19,118.16,117.53,77.56,76.71,65.34, 45.02,44.04,42.95,40.41,39.24,38.12,36.19,34.49,32.98,31.48,30.63,29.87,28.55,22.60, 21.36,18.43,10.28.esi-ms[m-br]
:815.4.
[0076]
实施例9:cel溴代间苯芳香烃中间体的制备
[0077][0078]
135mg(0.3mmol)cel溶于15ml n,n-二甲基甲酰胺中,加入1,3-二(溴甲基)苯158 mg(0.6mmol)、nahco
3 125mg(1.2mmol),60℃搅拌反应8h,加入15ml水稀释,用二氯 甲烷萃取三次(每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥, 过滤,浓缩有机相,硅胶(200-300目)柱层析(洗脱剂为石油醚:乙酸乙酯=20:1v/v),真空干燥 过夜,得到cel溴代间苯芳香烃中间体(橘红色粉末,138mg,产率72.3%)。
[0079]1h nmr(300mhz,dmso,tms),δ7.46(1h,s),7.24(3h,m),7.06(1h,d,j=7.16hz), 6.99(1h,s),6.44(1h,s),6.39(1h,d,j=7.12hz),4.13(2h,m),3.72(2h,m),2.24(3h,s),1.43 (3h,s),1.27(3h,s),1.12(3h,s),0.98(3h,s),0.43(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ178.52,164.81,135.14,133.78,130.69,118.27,117.54,111.97,77.48,76.63,45.66,44.18, 40.58,39.39,38.26,36.27,34.79,33.73,31.65,30.86,29.71,21.64,18.77,10.55.esi-ms[m-br]
: 633.6.
[0080]
实施例10:化合物i-5的制备
[0081][0082]
127mg(0.2mmol)cel溴代间苯芳香烃中间体溶于15ml n,n-二甲基甲酰胺中,加入三 苯基膦82mg(0.24mmol),60℃搅拌反应24h后,加入15ml水稀释,用二氯甲烷萃取三次 (每次30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有 机相,硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合 物i-5(橘红色粉末,102mg,产率56.9%)。
[0083]1h nmr(300mhz,dmso,tms)δ7.78(15h,m),7.46(1h,s),7.24(3h,m),7.06(1h,d,j =7.14hz),6.97(1h,s),6.42(1h,s),6.37(1h,d,j=7.12hz),4.26(2h,m),3.94(2h,m),2.22 (3h,s),1.42(3h,s),1.28(3h,s),1.12(3h,s),0.96(3h,s),0.42(3h,s).
13
c nmr(75mhz, dmso,tms),δppm:δ178.18,177.86,171.35,165.45,165.36,145.98,136.17,135.19,134.55, 131.94,130.32,128.87,127.12,119.43,118.31,117.58,77.52,76.64,65.33,45.12,44.24,42.84, 40.34,39.64,38.41,36.37,34.56,32.98,31.41,30.66,29.88,28.67,22.75,21.46,18.48,10.33. esi-ms[m-br]
:815.4.
[0084]
实施例11:化合物
ⅱ‑
1的制备
[0085][0086]
114mg(0.2mmol)cel溴代丙烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三环己基 膦87mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物
ⅱ‑
1 (橘红色粉末,92mg,产率53.9%)。
[0087]1h nmr(300mhz,dmso,tms)δ7.06(1h,d,j=7.14hz),6.97(1h,s),6.53(1h,s), 6.37(1h,d,j=7.12hz),4.16(2h,m),3.58(2h,m),2.33(3h,s),2.10-1.61(30h,m),1.56(3h,s), 1.35(3h,s),1.21(3h,s),0.94(3h,s),0.53(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ188.48,171.68,168.15,146.48,139.75,133.63,121.86,118.38,117.49,105.52,77.48,76.82, 70.33,61.22,44.90,43.47,41.85,39.74,39.03,37.34,35.32,33.67,32.63,31.63,30.64,29.77, 28.96,27.29,26.89,25.72,22.87,21.78,20.85,19.11,17.70,10.49.esi-ms[m-br]
:771.5.
[0088]
实施例12:化合物
ⅱ‑
2的制备
[0089][0090]
117mg(0.2mmol)cel溴代丁烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三环己基 膦87mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物
ⅱ‑
2 (橘红色粉末,99mg,产率57.1%)。
[0091]1h nmr(300mhz,dmso,tms)δ7.09(1h,d,j=7.16hz),6.94(1h,s),6.51(1h,s), 6.35(1h,d,j=7.12hz),4.12(2h,m),3.60(2h,m),2.31(3h,s),2.12-1.63(30h,m),1.55(3h,s), 1.33(3h,s),1.20(3h,s),0.95(3h,s),0.51(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ189.13,171.84,168.35,146.71,139.94,133.81,121.86,118.66,117.85,105.74,77.87,76.82, 70.55,61.54,44.90,43.79,41.85,39.84,39.03,37.59,35.47,33.66,32.87,31.88,30.74,29.85, 28.96,27.52,26.89,25.92,22.87,21.88,20.91,19.43,17.82,10.57.esi-ms[m-br]
:785.6.
[0092]
实施例13:化合物
ⅱ‑
3的制备
[0093][0094]
120mg(0.2mmol)cel溴代戊烷中间体溶于15ml n,n-二甲基甲酰胺中,加入三环己基 膦87mg(0.24mmol),60℃搅拌反应24h,加入15ml水稀释,用二氯甲烷萃取三次(每次 30ml),合并有机层,饱和食盐水溶液(100ml)洗涤,无水硫酸钠干燥,过滤,浓缩有机相, 硅胶(200-300目)柱层析(洗脱剂为二氯甲烷:甲醇=50:1v/v),真空干燥过夜,得到化合物
ⅱ‑ꢀ
3(橘红色粉末,106mg,产率60.3%)。
[0095]1h nmr(300mhz,dmso,tms)δ7.07(1h,d,j=7.14hz),6.98(1h,s),6.54(1h,s), 6.37(1h,d,j=7.12hz),4.18(2h,m),3.59(2h,m),2.33(3h,s),2.13-1.65(30h,m),1.55(3h,s), 1.36(3h,s),1.22(3h,s),0.95(3h,s),0.52(3h,s).
13
c nmr(75mhz,dmso,tms),δppm:δ188.53,171.74,168.41,146.52,139.84,133.71,121.92,118.55,117.64,105.71,77.53,76.62, 70.56,61.46,44.82,43.54,41.64,39.53,39.13,37.59,35.47,33.77,
nmr(75m hz,dmso,tms),δppm:δ188.54,177.77,171.65,168.54,146.59,139.85,133.77,121.93, 119.88,119.66,118.54,117.74,105.73,77.58,76.69,70.57,61.49,44.81,43.59,41.68,39.58,39.22, 37.63,35.51,33.66,32.65,31.82,30.32,29.55,28.77,27.38,26.75,25.64,22.52,21.57,20.58, 19.23,17.67,10.57.esi-ms[m-br]
:833.6.
[0104]
实施例16:体外抗肿瘤活性研究
[0105]
采用四甲基氮唑蓝比色法(mtt法)对本发明雷公藤红素衍生物进行抗肿瘤活性测试,选 取雷公藤红素(cel)作为阳性对照药。
[0106]
仪器:超净工作台(sw-cj-1fd,airtech,苏净安泰)、恒温co2培养箱(3111, thermo,美国)、倒置生物显微镜(ix71,olympus,日本)、酶联免疫检测仪 (model680,bio-rad,美国)、平板摇床(kylin-bell lab instruments)、高压灭菌锅 (yxo.sg41.280,上海华线),离心机(sigma)。
[0107]
试剂:dmem(gibco)、胎牛血清(gibco)、胰蛋白酶(sigma),dmso (sigma)。
[0108]
细胞株:人非小细胞肺癌细胞株a549,人结肠癌细胞株hct116,人乳腺癌细胞株 mcf-7(均购自江苏凯基生物技术股份有限公司)。
[0109]
方法:将冻存的细胞株复苏,置于恒温37℃、co2培养箱中培养,每天换液一次,待其 处于指数生长期状态良好时即可铺板。加入1ml 0.25%胰蛋白酶消化液,消化1-2min,在 显微镜下观察细胞状态,当贴壁细胞变圆收缩时即可吸除消化液,加入1-2ml含10%胎牛 血清的dmem培养基制成细胞悬液,进行细胞计数,按照每孔5
×
104个细胞数及总孔数计 算所需细胞悬液的量,将此细胞悬液接种于96孔板上,100μl/孔,周围用pbs液封,置于 恒温37℃、co2培养箱中培养24h。用含10%胎牛血清的dmem培养基分别配制受试药物 (雷公藤红素衍生物)、阳性对照药(雷公藤红素)溶液成浓度梯度,加入到96孔板中,每个药 物3个复孔,加入等体积dmso作为空白对照组,培养48小时。将mtt试剂加入到96孔 板中,10μl/孔,继续孵育4h。吸除板内培养基,每孔加入100μl dmso,平板摇床振摇10 min使结晶溶解。用酶联免疫检测仪在波长570nm处检测每孔的吸光值,并按下列公式计 算细胞抑制率。3次初筛结果平均值为其最终抑制率,最终根据抑制率计算受试药物的ic
50
值(graphpad软件计算),3次重复实验结果为所测化合物的最终ic
50
值。
[0110]
细胞抑制率%=[(空白对照od值-给药组od值)/空白对照组od值]
×
100%
[0111]
表1:化合物对a549、hct116和mcf-7细胞株的抑制作用
[0112][0113]
综上所述,本发明雷公藤红素衍生物对a549、hct-116-116和mcf-7的细胞活性明显 高于雷公藤红素,且随着饱和烷基链的增长,活性更显著。以化合物
ⅱ‑
3活性最佳,其对 a549细胞株的ic
50
值为0.39
±
0.02μm(约提高了4倍),有望成为新的抗肿瘤候选药物, 值得进一步研究。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。