1.本发明属于天然药物及药物化学领域,涉及一类色原酮氮芥衍生物与抗肿瘤应用,具体涉及一系列具有抗肿瘤活性的色原酮氮芥衍生物的制备方法及其在抗肿瘤方面的用途。
背景技术:
2.色原酮骨架是大量生物活性分子的重要组成部分,同时也是许多天然产物、先导化合物和临床药物的核心部分。色原酮通过合成可得到结构多样的衍生物,可作为结构修饰的先导化合物,合成多种具有不同药理活性的衍生物。目前,色原酮及其衍生物已成为具有抗癌活性最重要的合成类化合物之一,国内外对色原酮的合成以及结构修饰研究活跃。
3.氮芥为一类dna烷化剂,现已被开发成多种具有广谱抗肿瘤活性的化疗药。但因其对癌细胞具有非特异性,往往需要大剂量的使用以起到杀死癌细胞的效果。而大剂量氮芥的使用会引起严重的副作用以及药物耐药性,限制其在临床上的进一步应用。因此很多研究者对氮芥进行结构修饰,为了获得活性更强、毒性更低、选择性更好的抗肿瘤候选化合物。
技术实现要素:
4.本发明要解决的技术问题是寻找抗肿瘤活性好的色原酮氮芥衍生物及其药学上可接受的盐,并进一步提供一种药物组合物。
5.为解决上述技术问题,本发明提供如下技术方案:
6.一种色原酮氮芥衍生物及其药学上可接受的盐,具有如下i的结构通式:
[0007][0008]
其中,r为含有1
‑
8个碳原子的烷基或含有1
‑
8个碳原子的烷氧基;杂原子x为n、o、s或se。
[0009]
优选地,r为含有1
‑
6个碳原子的烷基或含有1
‑
6个碳原子的烷氧基;杂原子x为n、o或s。
[0010]
更优选地,r为含有1
‑
4个碳原子的烷基或含有1
‑
4个碳原子的烷氧基;杂原子x为n或o。
[0011]
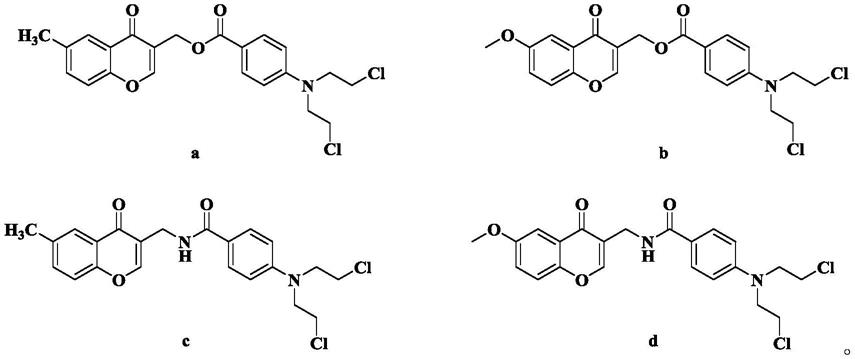
进一步地,本发明优选如下衍生物及其药学上可接受的盐结构式如a~d所示:
[0012][0013]
本发明上述的衍生物可用下列方法制备得到:
[0014][0015][0016]
(1)化合物1a
‑
b在n,n
‑
二甲基甲酰胺中与三氯氧磷室温反应得中间体2a
‑
b;然后将中间体2a
‑
b溶解于异丙醇中,加入碱性al2o3,回流反应得到化合物3a
‑
b;再将化合物3a
‑
b溶于二氯甲烷中,冰浴条件下滴加三溴化磷,后转移至室温反应,经处理得到的固体溶解于n,n
‑
二甲基甲酰胺中,加入氨水,室温反应得到化合物4a
‑
b;
[0017]
(2)化合物5在乙酸水溶液中,与环氧乙烷反应得到中间体6;中间体6先与三氯氧磷反应,而后在10%盐酸水溶液条件下得到化合物7。
[0018]
(3)将化合物3a
‑
b或4a
‑
b溶于无水二氯甲烷,依次加入edci、dmap或hobt,与化合物7室温反应得到目标化合物8a
‑
d。
[0019]
一种药物组合物,所述药物组合物含有治疗有效量的所述的通式i所示的色原酮氮芥衍生物及其药学上可接受的盐和药学上可接受的载体。
[0020]
上述通式i所示的色原酮氮芥衍生物及其药学上可接受的盐在制备治疗肿瘤疾病
的药物中的应用。
[0021]
进一步地,所述的肿瘤为乳腺癌肿瘤或肝癌肿瘤。
[0022]
上述药物组合物在制备治疗肿瘤疾病的药物中的应用。
[0023]
进一步地,所述的肿瘤为乳腺癌肿瘤或肝癌肿瘤。
[0024]
本发明以色原酮为先导化合物,设计并合成了一系列色原酮氮芥衍生物,并测试了合成衍生物在抗肿瘤方面的生物活性。
[0025]
药理试验证明,本发明的色原酮氮芥衍生物具有很好的抗肿瘤细胞增殖作用,可以用于进一步制备抗肿瘤药物。
具体实施方式
[0026]
下述非限定性实施例可以使本领域的普通技术人员更全面地理解本发明,但不以任何方式限制本发明。
[0027]
本发明实施例的衍生物合成路线如下:
[0028][0029]
实施例1
[0030][0031]
(1)将500mg化合物1a(3.33mmol)溶解于10ml dmf中,然后逐滴加入630μl三氯氧磷(6.72mmol),室温反应12h。tlc监测,反应基本完全,加入15ml水,有结晶析出,抽滤,烘干,得中间体2a 402.4mg。将50mg中间体2a(0.27mmol)溶解于10ml异丙醇中,加入1g碱性
al2o3(9.80mmol),然后75℃回流反应5h。tlc监测,反应完全,抽滤,将滤液浓缩得粗品,使用硅胶柱色谱(dcm:meoh)分离得到化合物3a(r=
‑
ch3)。
[0032]
(2)将5ml环氧乙烷(0.10mol)加入到1.53g 4
‑
氨基苯甲酸乙酯5(9.26mmol)溶解于12ml 65%乙酸水溶液的悬浮液中,在室温下搅拌24h。反应完全后,用乙酸乙酯萃取3次,饱和食盐水洗1次,无水硫酸钠干燥,过滤,浓缩,得中间体6 1.64g。在冰浴条件下慢慢滴加2ml pocl3于1.5g中间体6(6.00mmol)中,在50℃油浴反应0.5h。后加入3ml 10%盐酸水溶液室温反应12h,过滤,洗涤滤渣,真空浓缩得到化合物7。
[0033]
(3)将50mg化合物3a(0.26mmol)溶解在5ml无水二氯甲烷中,加入151.6mg edci(0.80mmol)、16.1mg dmap(0.13mmol)、68.9mg化合物7(0.21mmol)并在室温下反应12h。反应完成后,将反应液倒入水中,二氯甲烷萃取3次,饱和食盐水洗1次,无水na2so4干燥,浓缩得粗品。硅胶柱色谱(dcm:meoh)分离得到目标化合物8a。白色油状,产率66.5%。1h nmr(cdcl3,400mhz),δ:8.12(1h,s,2
‑
h),8.02(1h,d,j=2.0hz,5
‑
h),7.94(2h,d,j=9.1hz,ar
‑
h),7.47(1h,dd,j=8.6,2.0hz,7
‑
h),7.34(1h,d,j=8.6hz,8
‑
h),6.63(2h,d,j=9.1hz,ar
‑
h),5.27(2h,s,
‑
ch2),3.78(4h,t,j=7.0hz,
‑
ch2),3.64(4h,t,j=7.0hz,
‑
ch2),2.45(3h,s,
‑
ch3);
13
c nmr(cdcl3,100mhz),δ:176.76,166.40,155.42,154.75,149.83,135.36,135.07,131.98(2),125.20,123.75,119.77,118.59,117.95,110.86(2),58.12,53.27(2),40.20(2),20.95;hrms(esi)m/z calcd for c
22
h
20
cl2no4[m
‑
h]
‑
432.0769,found 432.0748。
[0034]
实施例2
[0035][0036]
实施例1步骤(1)中合成3a的步骤替换成:将553mg化合物1b(3.33mmol)溶解于10ml dmf中,然后逐滴加入630μl三氯氧磷(6.72mmol),室温反应12h。tlc监测,反应基本完全,加入15ml水,有结晶析出,抽滤,烘干,得中间体2b 410mg。将55mg中间体2b(0.27mmol)溶解于10ml异丙醇中,加入1g碱性al2o3(9.80mmol),然后75℃回流反应5h。tlc监测,反应完全,抽滤,将滤液浓缩得粗品,使用硅胶柱色谱(dcm:meoh)分离得到化合物3b(r=
‑
och3)。
[0037]
其余步骤参照实施例1的合成方法制备得化合物8b,黄色油状,产率43.8%。1h nmr(cdcl3,400mhz),δ:8.14(1h,s,2
‑
h),7.95(2h,d,j=9.1hz,ar
‑
h),7.61(1h,d,j=3.1hz,5
‑
h),7.40(1h,d,j=9.1hz,8
‑
h),7.26(1h,dd,j=9.1,3.1hz,7
‑
h),6.64(2h,d,j=9.1hz,ar
‑
h),5.27(2h,s,
‑
ch2),3.90(3h,s,
‑
och3),3.79(4h,t,j=6.8hz,
‑
ch2),3.64(4h,t,j=6.8hz,
‑
ch2);
13
c nmr(cdcl3,100mhz),δ:176.56,166.41,157.08,155.32,151.34,149.83,132.00(2),124.70,123.95,119.62,119.18,118.62,110.86(2),104.95,58.13,55.95,53.29(2),40.10(2);hrms(esi)m/z calcd for c
22
h
20
cl2no5[m
‑
h]
‑
448.0719,found 448.0714。
[0038]
实施例3
[0039][0040]
实施例1步骤(1)中合成3a的步骤替换成:将500mg化合物1a(3.33mmol)溶解于10ml dmf中,然后逐滴加入630μl三氯氧磷(6.72mmol),室温反应12h。tlc监测,反应基本完全,加入15ml水,有结晶析出,抽滤,烘干,得中间体2a 402.4mg。将50mg中间体2a(0.27mmol)溶解于10ml异丙醇中,加入1g碱性al2o3(9.80mmol),然后75℃回流反应5h。tlc监测,反应完全,抽滤,将滤液浓缩得粗品,使用硅胶柱色谱(dcm:meoh)分离得到化合物3a 15mg。将500mg化合物3a(2.62mmol)溶解于10ml dcm中,冰浴下滴加750μl三溴化磷(7.90mmol),反应10min,转移至室温反应15h。tlc监测,反应完全,二氯甲烷萃取3次,饱和食盐水洗1次,无水na2so4干燥,过滤,浓缩,得652mg黄色固体。所得固体用10ml dmf溶解,加入10ml氨水,室温反应过夜。tlc监测,反应完全,乙酸乙酯萃取3次,饱和食盐水洗1次,无水na2so4干燥,过滤,浓缩得粗品。经硅胶柱色谱(dcm:meoh)分离,得化合物4a(r=
‑
ch3)。
[0041]
实施例1步骤(3)中合成8a的步骤替换成:将26.4mg化合物4a(0.14mmol)溶解在4ml无水二氯甲烷中,加入39.9mg edci(0.21mmol)、22.5mg hobt(0.17mmol)、36.4mg化合物7(0.14mmol)并在室温下反应6h。反应完成后,将反应液倒入水中,二氯甲烷萃取3次,饱和食盐水洗1次,无水na2so4干燥,浓缩得粗品。硅胶柱色谱(dcm:meoh)分离得到目标化合物8c。
[0042]
其余步骤参照实施例1,制备得化合物8c,白色固体,产率36.8%。1h nmr(cdcl3,400mhz),δ:8.17(1h,s,2
‑
h),7.98(1h,d,j=2.0hz,5
‑
h),7.70(2h,d,j=8.9hz,ar
‑
h),7.48(1h,dd,j=8.6,2.0hz,7
‑
h),7.36(1h,d,j=8.6hz,8
‑
h),7.14(1h,s,
‑
nh),6.64(2h,d,j=8.9hz,ar
‑
h),4.45(2h,d,j=5.6hz,
‑
ch2),3.76(4h,m,
‑
ch2),3.62(4h,m,
‑
ch2),2.45(3h,s,
‑
ch3);
13
c nmr(cdcl3,100mhz),δ:178.5,167.0,154.9,154.5,148.7,135.3,135.2,129.1(2),124.8(2),123.6,121.0,118.1,111.1(2),53.3(2),40.2(2),36.3,20.9;hrms(esi)m/z calcd for c
22
h
21
cl2n2o3[m
‑
h]
‑
431.0929,found 431.0921。
[0043]
实施例4
[0044][0045]
实施例1步骤(1)中合成3a的步骤替换成:将553mg化合物1b(3.33mmol)溶解于10ml dmf中,然后逐滴加入630μl三氯氧磷(6.72mmol),室温反应12h。tlc监测,反应基本完全,加入15ml水,有结晶析出,抽滤,烘干,得中间体2b 410mg。将55mg中间体2b(0.27mmol)溶解于10ml异丙醇中,加入1g碱性al2o3(9.80mmol),然后75℃回流反应5h。tlc监测,反应完全,抽滤,将滤液浓缩得粗品,使用硅胶柱色谱(dcm:meoh)分离得到化合物3b 22mg。将
540mg化合物3b(2.62mmol)溶解于10ml dcm中,冰浴下滴加750μl三溴化磷(7.90mmol),反应10min,转移至室温反应15h。tlc监测,反应完全,二氯甲烷萃取3次,饱和食盐水洗1次,无水na2so4干燥,过滤,浓缩,得681mg黄色固体。所得固体用10ml dmf溶解,加入10ml氨水,室温反应过夜。tlc监测,反应完全,乙酸乙酯萃取3次,饱和食盐水洗1次,无水na2so4干燥,过滤,浓缩得粗品。经硅胶柱色谱(dcm:meoh)分离,得化合物4b(r=
‑
och3)。
[0046]
实施例1步骤(3)中合成8a的步骤替换成:将28.7mg化合物4b(0.14mmol)溶解在4ml无水二氯甲烷中,加入39.9mg edci(0.21mmol)、22.5mg hobt(0.17mmol)、36.4mg化合物7(0.14mmol)并在室温下反应6h。反应完成后,将反应液倒入水中,二氯甲烷萃取3次,饱和食盐水洗1次,无水na2so4干燥,浓缩得粗品。硅胶柱色谱(dcm:meoh)分离得到目标化合物8d。
[0047]
其余步骤参照实施例1,制备得化合物8d,白色固体,产率43.5%。1h nmr(cdcl3,400mhz),δ:8.17(1h,s,2
‑
h),7.70(2h,d,j=8.8hz,ar
‑
h),7.55(1h,d,j=3.1hz,5
‑
h),7.40(1h,d,j=9.1hz,8
‑
h),7.27(1h,dd,j=9.1,3.1hz,7
‑
h),7.11(1h,s,
‑
nh),6.64(2h,d,j=8.8hz,ar
‑
h),4.46(2h,d,j=5.7hz,
‑
ch2),3.89(3h,d,
‑
och3),3.76(4h,m,
‑
ch2),3.62(4h,m,
‑
ch2);
13
c nmr(cdcl3,100mhz),δ:178.3,167.0,157.0,154.4,151.6,148.7,129.1(2),124.6,124.1,120.4,119.8,111.1(2),104.5,55.9,53.3(2),40.1(2),36.3,29.7;hrms(esi)m/z calcd for c
22
h
21
cl2n2o4[m
‑
h]
‑
447.0878,found 447.0878。
[0048]
实施例5
[0049]
下面是本发明部分化合物的药理实验结果:
[0050]
实验设备与试剂
[0051]
仪器超净工作台(苏净集团安泰公司)
[0052]
恒温培养箱(thermo electron corporation)
[0053]
酶标仪(bio
‑
rad公司)
[0054]
倒置生物显微镜(重庆光学仪器厂)
[0055]
试剂细胞培养基rpmi
‑
1640、dmem(高糖)(gibco公司)胎牛血清(杭州四季清有限公司)
[0056]
cck
‑
8(biosharp公司产品)
[0057]
dmso(sigma公司)
[0058]
细胞株人乳腺癌细胞mcf
‑
7、人乳腺癌细胞mda
‑
mb
‑
231、人肝癌细胞hepg2、bel
‑
7402
[0059]
实验方法
[0060]
细胞抑制活性实验方法
[0061]
细胞在37℃、5%co2饱和湿度的培养箱中常规培养。培养液为含10%热灭活胎牛血清,青霉素100u/ml和链霉素100u/ml的高糖dmem细胞培养基。48h更换培养液,细胞贴壁后,用0.25%胰蛋白酶消化传代。实验用细胞均处于对数生长期,cck
‑
8法表明细胞活力>95%。
[0062]
取处于对数生长期状态良好的细胞一瓶,加入消化液(0.125%胰蛋白酶 0.01%edta)消化,计数2~4
×
104cell/ml,制成细胞悬液接种于96孔板上,100μl/孔,置恒温co2培养箱中培养24小时。换液,加入受试药物,100μl/孔,培养72小时。将cck
‑
8加入96孔板中,50
μl/孔,培养箱中孵育4小时。吸去上清液,加dmso,200μl/孔,平板摇床上震荡10分钟。受试物考察0.001至100μm以十倍浓度递增的6个浓度,用酶联免疫监测仪在波长为450nm处测定每孔的吸光度,分别计算各浓度下的细胞抑制率。
[0063]
抑制率计算方法:
[0064][0065]
药敏孔相对od值=药敏孔绝对od值﹣空白对照孔绝对od值
[0066]
实验结果
[0067]
表1实施例对2种人乳腺癌和1种人肝癌细胞株抗增殖活性的ic
50
值(μm)
[0068][0069]
药理试验证明,本发明的目标衍生物具有更好的抗乳腺癌和肝癌细胞增殖活性,可以用于进一步制备抗肿瘤药物。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。