1.本发明涉及有机合成技术领域,具体领域为一种阿托伐他汀钙中间体的制备方法。
背景技术:
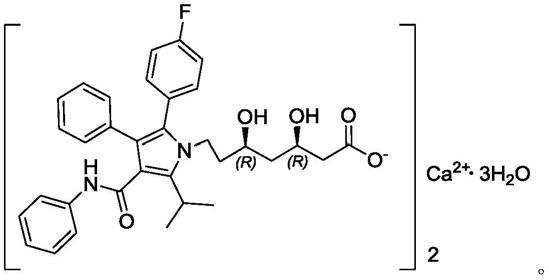
2.阿托伐他汀钙,化学名称为[r
‑
(r’,r’)]
‑2‑
(4
‑
氟苯基)
‑
β,α
‑
二羟基
‑5‑
(1
‑
甲基乙基)
‑3‑
苯基
‑4‑
[(苯胺基)羰基]
‑1‑
氢
‑
吡咯
‑1‑
庚酸钙盐(2:1)三水合物,cas号是134523
‑
03
‑
8,是hmg
‑
coa还原酶的选择性、竞争性抑制剂,可治疗总胆固醇升高、低密度脂蛋白胆固醇升高、载脂蛋白b升高和甘油三酯升高。其结构式如下:
[0003][0004]
阿托伐他汀钙的合成涉及两个重要的中间体,即n
‑
苯基异丁酰乙酰胺(以下简称“化合物1”)和4
‑
(4
‑
氟苯基)
‑2‑
(2
‑
甲基丙酰基)
‑3‑
苯基
‑4‑
氧代
‑
n
‑
苯基丁酰胺(以下简称“m4”)。
[0005]
n
‑
苯基异丁酰乙酰胺,cas号是124401
‑
38
‑
3,是制备阿托伐他汀钙的关键中间体,其结构式是
[0006]
中国专利cn101307009a公开了一种n
‑
苯基异丁酰乙酰胺的合成工艺,以甲苯为反应溶剂,将甲基异丙基酮滴加至碳酸二甲酯中,在氢化钠的作用下反应4h,得到的中间体再与苯胺继续反应2h,操作步骤简单,但制得n
‑
苯基异丁酰乙酰胺收率只有76%。
[0007]
中国专利cn101337906b报道了一种n
‑
苯基异丁酰乙酰胺的制备方法,以异丁酰乙酸甲酯和苯胺为原料,在4
‑
二甲氨基吡啶的作用下进行酰胺化反应,后处理使用石油醚、水、盐酸混合溶剂结晶纯化,合成n
‑
苯基异丁酰乙酰胺。收率虽然高达98%,但是石油醚是闪点低且易燃易爆易挥发的液体,毒害大,操作安全性低。
[0008]
美国专利us2004072893a公开了一种m4的合成工艺路线,以异丁酰氯、米氏酸、苯胺为起始原料合成n
‑
苯基异丁酰乙酰胺,再经过aldol缩合反应制得α,β
‑
不饱和酮,最后加入4
‑
氟苯甲醛得到m4。该工艺的原料廉价易得,但第一步多组分反应的收率不高;其次,工艺过程中使用剧毒试剂nacn,具有较高的安全风险,且废水污染严重。
[0009]
技术实现要素:
[0010]
本发明的目的在于提供一种无毒无害、收率高、纯度高、减少三废排放的阿托伐他汀钙中间体的制备方法。
[0011]
为实现上述目的,本发明提供如下技术方案:
[0012]
一种阿托伐他汀钙中间体的制备方法:以苯胺和双乙烯酮为起始原料,在有机溶剂中发生酰化反应,得到n
‑
乙酰乙酰苯胺(化合物2),化合物2再与异丁酰氯反应得到2
‑
乙酰基
‑4‑
甲基
‑3‑
氧代
‑
n
‑
苯基戊酰胺(化合物6),2
‑
乙酰基
‑4‑
甲基
‑3‑
氧代
‑
n
‑
苯基戊酰胺(化合物6)与氯化铵水溶液反应合成n
‑
苯基异丁酰乙酰胺,再与2
‑
卤代
‑1‑
(4
‑
氟苯基)
‑2‑
苯乙酮(化合物m3)反应,合成阿托伐他汀钙中间体(化合物m4),其反应路线如下:
[0013][0014][0015]
具体为:
[0016]
(1)制备n
‑
乙酰乙酰苯胺(化合物2)
[0017]
在反应釜中加入有机溶剂,设置温度10~40℃,边搅拌边同时滴加双乙烯酮和苯胺,滴加完毕后开始发生酰化反应;反应完成后冷却至0℃,过滤,烘干,得到化合物2;
[0018]
有机溶剂是乙醇、甲醇、丙酮、甲苯、二氯甲烷、氯仿、四氢呋喃其中的任一种,优选为乙醇;
[0019]
苯胺和双乙烯酮的投料摩尔比为1:0.8~1.8,优选为1:1,可以但不局限于1:0.8、1:1、1:1.2、1:1.4、1:1.6、1:1.8;
[0020]
苯胺和有机溶剂的投料质量比是1:1~5,优选为1:3.5;
[0021]
酰化反应温度是10~40℃,优选为30℃,可以但不局限于10℃、15℃、20℃、25℃、30℃、35℃、40℃;
[0022]
酰化反应时间是1~4h,优选为3h,可以但不局限于1h、1.5h、2h、2.5h、3h、3.5h、4h。
[0023]
(2)制备n
‑
苯基异丁酰乙酰胺(化合物1)
[0024]
向反应釜中加入化合物2、4
‑
二甲氨基吡啶(dmap)、甲苯,搅拌使其混合均匀,再加入氢氧化钙、氧化钙;降温至0~10℃滴加异丁酰氯,升温进行第一次反应;控制温度,滴加氯化铵/水溶液,进行第二次反应,至反应终点。
[0025]
对反应后的体系进行分液、洗涤、浓缩、干燥、重结晶、离心,得到化合物1。
[0026]
化合物2和异丁酰氯的投料摩尔比是1:0.5~2.5,优选为1:1.2,可以但不局限于1:0.5、1:0.7、1:1、1:1.2、1:1.5、1:1.8、1:2、1:2.2、1:2.5;
[0027]
化合物2与4
‑
二甲氨基吡啶的投料摩尔比是1:0.001~0.01,优选为1:0.002,可以但不局限于1:0.001、1:0.002、1:0.004、1:0.005、1:0.007、1:0.009、1:0.01;
[0028]
步骤(2)中,第一次反应的温度是5~20℃,优选为15℃,可以但不限于5℃、10℃、15℃、20℃;第一次反应的时间是3~7h,优选为5h,可以但不局限于3h、3.5h、4h、5h、6h、7h;
[0029]
步骤(2)中,第二次反应的温度是15~35℃,优选为25℃,可以但不局限于15℃、20℃、25℃、30℃;第二次反应的时间是3~8h,优选为6h,可以但不局限于3h、3.5h、4h、4.5h、5h、6h、7h、8h。
[0030]
(3)制备阿托伐他汀钙中间体(化合物m4)
[0031]
向化合物1中加入异丙醇,搅拌均匀后加入碳酸钾和水,在25℃搅拌0.5h反应得到混合液1备用;
[0032]
将化合物m3溶于异丙醇,缓慢滴加至上述混合液1中,升温至45℃进行化学反应,中控显示原料反应完全时停止加热,冷至室温。
[0033]
对反应产物进行减压浓缩除去异丙醇,再析晶、抽滤、干燥、重结晶,得到阿托伐他汀钙中间体m4。
[0034]
反应时间是8~20h,优选为13h,可以但不限于8h、9h、10h、11h、12h、13h、14h、15h、16h、17h、18h、19h、20h。
[0035]
化合物1和化合物m3的投料摩尔比是1:0.5~4,优选为1:1,可以但不限于1:0.5、1:0.8、1:1、1:1.2、1:1.5、1:1.8、1:2、1:2.3、1:2.5、1:2.7、1:3、1:3.3、1:3.5、1:3.8、1:4。
[0036]
与现有技术相比,本发明的有益效果是:
[0037]
本发明制备方法以苯胺、双乙烯酮、异丁酰氯为原料,合成目标产物,操作简单,成本较低,为相关研究提供了借鉴经验;本方法通过简单的分液、洗涤、重结晶进行后处理,避免使用毒害较大的石油醚和氰化钠,操作方法简单,操作安全性高,产品纯度高、收率高,对环境友好。
具体实施方式
[0038]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0039]
实施例1
[0040]
一种阿托伐他汀钙中间体的制备方法,具体步骤为:
[0041]
(1)制备n
‑
乙酰乙酰苯胺(化合物2)
[0042][0043]
在反应釜中加入乙醇(325g),设置温度30℃,一边搅拌一边同时滴加双乙烯酮(84g,1mol)和苯胺(93g,1mol),滴加完毕后持续搅拌,在30℃开始发生酰化反应3h;反应完成后冷却至0℃,过滤,烘干,得到白色粉末(177g,收率100%);
[0044]
(2)制备n
‑
苯基异丁酰乙酰胺(化合物1)
[0045][0046]
向反应釜中加入化合物2(177g,1mol)、4
‑
二甲氨基吡啶(0.24g,0.002mol)、甲苯(921g,10mol),搅拌使其混合均匀,得到澄清的浅黄色溶液;控制温度20~25℃,再加入氢氧化钙(111g,1.5mol)、氧化钙(5.6g,0.1mol);降温至5~8℃滴加异丁酰氯(128g,1.2mol),升温至15℃进行第一次反应5h,薄层色谱分析法监测化合物2反应完全;控制温度25℃,滴加(90g,1.7mol)氯化铵/721g水溶液,进行第二次反应6h,薄层色谱分析法监测化合物6反应完全。
[0047]
向体系中滴加3n盐酸,控制温度25℃,体系由悬浊状态变成澄清的两相,分液,取有机相,用饱和碳酸氢钠洗涤、分液,取有机相加水洗涤、分液,再取有机相,浓缩至干,得到化合物1粗品;向粗品中加入正己烷,搅拌充分后加入水,再搅拌均匀,会有大量固体析出,离心,干燥,得白色固体(191g,收率93.2%),纯度为99.6%。
[0048]
本发明对于反应中滴加的具体加入方式没有特殊限定,根据试剂生产规模,采用本领域技术人员熟知的加入方式进行滴加,并且保证混合液内温不超过设定温度
±
3℃即可。
[0049]
(3)制备阿托伐他汀钙中间体(化合物m4)
[0050][0051]
将化合物1(70,0.34mol)加入三口瓶中,再加入异丙醇(800ml),搅拌成均匀体系,一次性加入碳酸钾(47.2g),水(10g),25℃搅拌0.5小时(此时体系呈淡黄色浆状,使化合物1充分活化),用冰水使体系降温到5℃,得到混合液1;
[0052]
将化合物m3
‑
br(x=br,100g,0.34mol)溶于异丙醇(200ml)中,缓慢滴加到上述混合液1中,此时体系中析出大量白色固体,升温到45℃,反应13h;
[0053]
原料反应完全后,减压浓缩除去异丙醇,然后缓慢往体系中滴加约1000g水,充分搅拌,缓慢降温到0
‑
5℃搅拌3小时,充分析晶,抽滤,干燥后的固体采用m4:甲醇:水=1:4:1(w/v)重结晶,得到化合物m4,纯度99.6﹪,收率为98%。
[0054]
本发明对于滴加的具体加入方式没有特殊限定,根据试剂生产规模,采用本领域技术人员熟知的加入方式进行滴加即可。
[0055]
实施例2~11
[0056]
按照实施例1中公开的方式,仅分别改变第一步反应中投料比或其他条件,详见表1。
[0057]
注:表中空白部分代表该条件与实施例1中的相同。
[0058]
表1第一步反应的不同条件与结果
[0059][0060][0061]
实施例12~21
[0062]
按照实施例1中公开的方式,仅分别改变第二步反应中投料比或其他条件,详见表2。
[0063]
注:表中空白部分代表该条件与实施例1中的相同。
[0064]
表2第二步反应的不同条件与结果
[0065][0066]
其中,dmap表示4
‑
二甲氨基吡啶。
[0067]
实施例22~26
[0068]
按照实施例1中公开的方式,仅分别改变第三步反应中投料比或其他条件,详见表3。
[0069]
注:表中空白部分代表该条件与实施例1中的相同。
[0070]
表3第三步反应的不同条件与结果
[0071][0072]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。