3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼及其甲基
‑
d3氘代形式的合成
发明领域
1.本发明涉及化学合成领域,提供了化合物(i)或其盐的合成方法:
[0002][0003]
其中r1代表甲基或甲基
‑
d3,因此对应于3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼或其甲基
‑
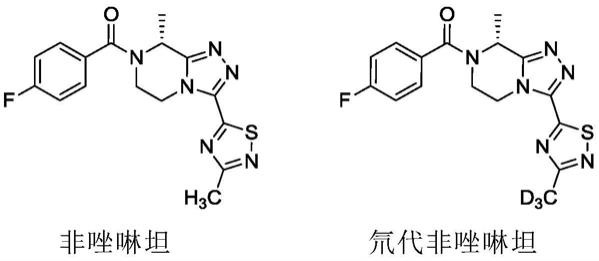
d3氘代形式。这些化合物是合成药物化合物的关键中间体,特别是非唑啉坦(fezolinetant)和氘代非唑啉坦。
[0004]
发明背景
[0005]
非唑尼坦是作为nk
‑
3受体的选择性拮抗剂而开发的,是一种有用的治疗化合物,特别是在治疗和/或预防性激素依赖性疾病方面。非唑啉坦对应于(r)
‑
(4
‑
氟苯基)
‑
(8
‑
甲基
‑3‑
(3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
基)
‑
5,6
‑
二氢
‑
[1,2,4]三唑并[4,3
‑
a]吡嗪
‑
7(8h)
‑
基)甲酮,在wo2014/154895中有所描述。
[0006]
氘代非唑啉坦(r)
‑
(4
‑
氟苯基)
‑
(8
‑
甲基
‑3‑
(3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
基)
‑
5,6
‑
二氢
‑
[1,2,4]三唑并[4,3
‑
a]吡嗪
‑
7(8h)
‑
基)甲酮也是出于同样的目的开发的,在wo2019/012033中有所描述。
[0007][0008]
合成非唑啉坦和氘代非唑啉坦的方法在wo2014/154895和wo2019/012033中有所描述。这些方法涉及3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼或其氘代形式作为关键中间体(i):
[0009][0010]
其中r1代表甲基或甲基
‑
d3。
[0011]
wo2013/050424公开了非氘代中间体3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼(i
‑
1)的合成方法,它是由相应的甲酯(ii
‑1‑
a)制备的,该甲酯由乙酰胺、氯羰基亚磺酰氯和氰基甲酸甲酯一步制备:
[0012][0013]
然而,氯羰基亚磺酰氯和氰基甲酸甲酯是危险试剂,在大规模生产时会造成原料问题,对于大规模生产应当避免其使用。此外,这种合成路线会产生硫杂质,其对最终药物产品的质量产生不良影响。此外,即使经过优化,中间体(i
‑
1)的总产率仍低于30%。
[0014]
获得相应的氘代中间体3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼(i
‑
2)的方法与wo2019/012033中所描述的相同,其具有相同缺点。
[0015]
因此,需要一种更安全、稳固、可大规模化和更高效的中间体(i)合成方法。
[0016]
本文提供了一种由相应的酯(ii)合成中间体(i)的方法,该酯本身是通过对相应的卤化中间体(iii)进行烷氧羰基化获得的:
[0017][0018]
或其盐,其中r1代表甲基或甲基
‑
d3,r2代表烷基或芳烷基,x代表卤素原子。
[0019]
曾经报道过这种类型的烷氧羰基化反应发生在一些更稳固的5元杂环上,例如噻吩、呋喃、噻唑、咪唑、噁唑、吡唑和吲哚。然而,还从未报道过其发生在不那么稳固的杂环(即含有超过2个杂原子)上,其更容易开环,例如本发明中的噻二唑环。在形成1,2,4
‑
噻二唑的开放中间体的情况下,将产生反应性非常高的中间体(例如巯基脒)和挥发性副产物(特别是在r1是甲基的情况下)(类似的更稳固的噁唑参见:verrier等人,beilstein j.org.chem.,2011,7,1584
‑
1601;bellina等人,current org.chem.,2008,12(9),774
‑
790和strotman等人,org.lett.,2010,12,3578
‑
3581)。
[0020]
本发明方法的优点是不涉及特殊试剂的特别危险的化学。总产率非常令人满意(超过67%),即使是在大规模化后,正如下面的实验部分所证明的那样。
[0021]
式(iii)的起始卤化化合物可以由式(iv)的胺化合物通过sandmeyer反应获得:
[0022][0023]
本发明还提供了一种合成氘代中间体(iv)的新方法。该方法包括在氘代乙腈(vii
‑
2)上进行pinner反应,首先将其转化为pinner盐(vi
‑
2)(步骤a),之后转化成氘代乙脒(v
‑
2)(步骤b),之后形成噻二唑环(步骤c)以提供化合物(iv
‑
2):
[0024][0025]
该方法的优点是所产生的同位素纯度非常令人满意,下面的实验部分能证明这一点。
[0026]
本发明还提供了一种合成氘代中间体(iv)的替代新方法。该方法包括由氘代乙腈(vii
‑
2)形成氘代n
‑
羟基乙脒(ix
‑
2),之后用甲苯磺酰化将其活化形成中间体(x
‑
2),之后形成噻二唑环以提供化合物(iv
‑
2):
[0027][0028]
该方法也具有同位素纯度非常令人满意的优点。
[0029]
发明概述
[0030]
因此,本发明涉及一种制备式(i)化合物或其盐的方法:
[0031][0032]
其中r1代表甲基或甲基
‑
d3;
[0033]
所述方法包括以下步骤:
[0034]
a)对式(iii)化合物进行烷氧羰基化反应:
[0035][0036]
其中x代表卤素;r1代表甲基或甲基
‑
d3;
[0037]
获得式(ii)化合物:
[0038][0039]
其中r1代表甲基或甲基
‑
d3;r2代表烷基或芳烷基;
[0040]
以及
[0041]
b)通过使式(ii)化合物与一水合肼反应形成式(i)化合物。
[0042]
在一个实施方式中,在本发明的方法中,x代表溴或碘;优选地,x代表溴。在一个实施方式中,在本发明的方法中,r2代表甲基或乙基;优选地,r2代表乙基。
[0043]
在一个实施方式中,步骤a)的烷氧羰基化反应在一氧化碳、钯催化剂、碱和醇溶剂的存在下进行,并且任选在有机磷配体的存在下进行。
[0044]
在一个实施方式中,钯催化剂是pd(oac)2,有机磷配体是4,5
‑
双(二苯基膦)
‑
9,9
‑
二甲基氧杂蒽(xantphos),碱是乙酸钠,溶剂选自乙醇、甲醇、甲基叔丁基醚与乙醇或甲醇的混合物。优选地,步骤a)在乙醇作为溶剂或在乙醇与甲基叔丁基醚的混合物中进行。优选地,步骤a)在50℃
‑
150℃的温度下进行;优选在63℃
‑
67℃的温度下进行;更优选在约65℃的温度下进行。在一个实施方式中,步骤a)在fe(co)5的存在下进行。
[0045]
在另一个实施方式中,钯催化剂是双(三苯基膦)氯化钯(pd(pph3)2cl2),碱是三乙胺,溶剂是乙醇。
[0046]
在一个实施方式中,步骤a)的烷氧羰基化反应通过锂交换进行,首先使式(iii)化合物与有机锂试剂接触,之后加入氯甲酸酯或氰基甲酸酯。优选地,有机锂试剂是正己基锂,氯甲酸酯是氯甲酸烷基酯,优选氯甲酸乙酯。
[0047]
在一个实施方式中,本发明的方法包括对式(iv)化合物或其盐进行sandmeyer反应的预步骤:
[0048][0049]
其中r1代表甲基或甲基
‑
d3;
[0050]
从而形成式(iii)化合物。
[0051]
sandmeyer反应可以在亚硝酸钠和溴化氢的存在下在水性介质中进行。替代地,sandmeyer反应可以在叔丁基亚硝酸酯和碘的存在下或在碘化钾和对甲苯磺酸的存在下进行。
[0052]
本发明还涉及一种制备3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
胺(iv
‑
2)或其盐的方法:
[0053][0054]
其包括以下步骤:
[0055]
a)在hcl的存在下使d3
‑
乙腈与乙醇反应形成式(vi
‑
2)的pinner盐:
[0056][0057]
b)使pinner盐(vi
‑
2)与氨反应形成d3
‑
乙脒(v
‑
2)或其盐:
[0058][0059]
以及
[0060]
c1)使d3
‑
乙脒(v
‑
2)与溴、硫氰酸盐和甲醇钠反应以获得式(iv
‑
2)化合物;或
[0061]
c2)使d3
‑
乙脒(v
‑
2)首先与次氯酸钠(naocl)反应,之后与硫氰酸盐反应,以获得式(iv
‑
2)化合物。
[0062]
定义
[0063]
在本发明中,下列术语具有如下含义:
[0064]
‑“
约”,出现在数字之前,是指上下不超过该数字值的10%。
[0065]
‑“
醇溶剂”是指能够溶解溶质(化学上不同的液体、固体或气体)并形成溶液的醇,即包含至少一个与碳原子相连的羟基官能团(
‑
oh)的有机化合物。醇溶剂的实例包含甲醇、乙醇和异丙醇。
[0066]
‑“
烷氧羰基化反应”是指形成c
‑
c键的化学反应,其能够在链上引入烷氧羰基部分。“烷氧羰基部分”是指基团
‑
c(=o)
‑
o
‑
烷基,其中烷基定义为包括烷基本身被取代的情况,例如被芳基取代(即形成芳烷基),形成
‑
c(=o)
‑
o
‑
烷基
‑
芳基。
[0067]
‑“
烷基”是指式c
n
h
2n 1
的烃基,其中n是大于或等于1的数。通常,本发明的烷基包含
1
‑
6个碳原子,优选1
‑
4个碳原子。烷基可以是直链的或支链的,并且可以如本文所示被取代。烷基的实例有甲基、乙基、正丙基、异丙基、丁基及其异构体(如正丁基、异丁基和叔丁基)、戊基及其异构体、己基及其异构体。
[0068]
‑“
芳基”是指多不饱和的芳香族烃基,其具有一个单环(即苯基)或多个融合在一起的芳环(如萘),或共价连接的多个环,通常包含5
‑
12个原子,优选6
‑
10个原子,其中至少一个环是芳香族的。芳环可以任选地包括一至两个与之稠合的附加环(环烷基、杂环基或杂芳基)。芳基的非限定性实例包含苯基、联苯基、亚联苯基、萘
‑1‑
或
‑2‑
基。在一个实施方式中,芳基是苯基。任选地,芳基可以被一个或多个基团取代,例如烷氧基。在具体实施方式中,芳基被1
‑
3个烷氧基取代,优选被1
‑
3个甲氧基取代。
[0069]
‑“
芳烷基”是指芳基烷基的部分,其中芳基和烷基如本文所定义。芳烷基的实例包括苄基,4
‑
甲氧基苄基(pmb)、2,4
‑
二甲氧基苄基(dmb)和2,4,6
‑
三甲氧基苄基(tmb)。
[0070]
‑“
羰基化反应”是指形成c
‑
c键的化学反应,其能够在链上引入羰基部分,例如
‑
c(=o)
‑
o
‑
r2,其中r2例如是烷基或芳烷基。
[0071]
‑“
氯甲酸酯”是指式roc(o)cl的反应物,其中r例如可以代表烷基或芳烷基,分别形成烷基氯甲酸酯和芳烷基氯甲酸酯。
[0072]
‑“
氰基甲酸酯”是指式roc(o)cn的反应物,其中r例如可以代表烷基或芳烷基,分别形成烷基氰基甲酸酯和芳烷基氰基甲酸酯。
[0073]
‑“
卤素”或“卤族”是指氟、氯、溴或碘。在本发明中,优选的卤素基团是溴和碘。
[0074]
‑“
甲基
‑
d3”是指氘代部分
‑
cd3。
[0075]
‑“
有机锂试剂”是指含有碳
‑
锂键的金属有机化合物。有机锂试剂的实例包含正己基锂和正丁基锂。
[0076]
‑“
有机磷配体”是指含有磷的有机化合物,优选膦配体(phosphine ligands),即式pr3的化合物,其中r是有机衍生物。有机磷配体的实例包含三苯基膦(pph3)、三叔丁基膦(ptbu3)、4,5
‑
双(二苯基膦)
‑
9,9
‑
二甲基氧杂蒽(xantphos)、2,2'
‑
双(二苯基膦)
‑
1,1'
‑
联萘(binap)、1,3
‑
双(二苯基膦)丙烷(dppp)和二(1
‑
金刚烷基)
‑
正丁基膦
[0077]
‑“
钯催化剂”是指能够催化反应的钯配合物。钯催化剂的实例包括:乙酸钯(pd(oac)2)、氯化钯(pdcl2)、三(二亚苄基丙酮)二钯、双(二亚苄基丙酮)钯和双(三苯基膦)氯化钯(pd(pph3)2cl2)。钯催化剂包括原位活化的预催化剂,例如pd(pph3)2cl2,其在参与催化循环之前被还原为pd(0)配合物或被金属化成pd(ii)芳基配合物。
[0078]
‑“
pinner盐”是指腈与醇反应的产物,即亚氨基酯盐(烷基亚氨基酯盐)。
[0079]
‑“
sandmeyer反应”是指由芳基或杂芳基重氮盐通过自由基亲核芳香取代反应合成芳基或杂芳基卤化物的化学反应。
[0080]
‑“
硫氰酸盐”是指阴离子[scn]
‑
。它可以作为盐与诸如钾或钠等抗衡离子一起使用。
[0081]
‑“
甲苯磺酰基化”是指能够在羟基部分上引入对甲苯磺酰基(也称为甲苯磺酰基)的反应。在优选实施方式中,使用对甲苯磺酰氯进行甲苯磺酰基化。
具体实施方式
[0082]
化合物(i)的合成
[0083]
本发明涉及一种制备式(i)化合物或其盐的方法:
[0084][0085]
其中r1代表甲基或甲基
‑
d3;
[0086]
所述方法包括以下步骤:
[0087]
a)对式(iii)化合物进行烷氧羰基化反应:
[0088][0089]
其中x代表卤素;r1代表甲基或甲基
‑
d3;
[0090]
获得式(ii)化合物:
[0091][0092]
其中r1代表甲基或甲基
‑
d3;r2代表烷基或芳烷基;
[0093]
以及
[0094]
b)通过使式(ii)化合物与一水合肼反应形成式(i)化合物。
[0095]
本发明的方法中使用的最终化合物或中间体可以是盐的形式,包括酸加成盐和碱盐。在一个实施方式中,盐是药学上可接受的盐。合适的酸加成盐例如为乙酸盐、己二酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、碳酸氢盐/碳酸盐、硫酸氢盐/硫酸盐、硼酸盐、氨基磺酸盐、柠檬酸盐、环己胺磺酸盐、乙二磺酸盐、乙酰磺酸盐、甲酸盐、富马酸盐、葡萄糖酸盐、葡糖酸盐、葡萄糖醛酸盐、六氟磷酸盐、海苯酸盐、盐酸盐/氯化物、氢溴酸盐/溴化物、氢碘酸盐/碘化物、羟乙基磺酸盐、乳酸盐、苹果酸盐、马来酸盐、丙二酸盐、甲磺酸盐、萘磺酸盐、2
‑
萘磺酸盐、烟酸盐、硝酸盐、乳清酸盐、草酸盐、棕榈酸盐、双羟萘酸盐、磷酸盐/磷酸氢盐/磷酸二氢盐、焦谷氨酸盐、糖酸盐、硬脂酸盐、琥珀酸盐、鞣酸盐、酒石酸盐、甲苯磺酸盐、三氟乙酸盐和昔萘酸盐(xinofoate)。合适的碱盐例如有铝、精氨酸、苄星、钙、胆碱、二乙胺、二乙醇胺、甘氨酸、赖氨酸、镁、葡甲胺、乙醇胺、钾、钠、氨丁三醇、2
‑
(二乙氨基)乙醇、乙醇胺、吗啉、4
‑
(2
‑
羟乙基)吗啉和锌的盐。
[0096]
步骤a)
‑
烷氧羰基化
[0097]
在一个实施方式中,在式(iii)化合物中,x代表溴或碘。在一个优选实施方式中,在式(iii)化合物中,x代表溴。在另一个实施方式中,在式(iii)化合物中,x代表碘。
[0098]
在一个实施方式中,在式(ii)化合物中,r2代表c1
‑
c4烷基,优选甲基或乙基,更优选地,r2代表乙基。在另一个实施方式中,在式(ii)化合物中,r2代表芳烷基,优选苄基、4
‑
甲氧基苄基(pmb)、2,4
‑
二甲氧基苄基(dmb)或2,4,6
‑
三甲氧基苄基(tmb)。
[0099]
co/pd烷氧羰基化反应
[0100]
在一个实施方式中,步骤a)的烷氧羰基化反应是在一氧化碳的存在下进行的。在一个实施方式中,一氧化碳在1
‑
20巴,优选3.5
‑
8.5巴,更优选4
‑
5巴的压力下使用。
[0101]
在一个实施方式中,步骤a)的烷氧羰基化反应是在钯催化剂的存在下进行的。在一个实施方式中,钯催化剂例如选自乙酸钯(pd(oac)2)、氯化钯(pdcl2)、三(二亚苄基丙酮)二钯、双(二亚苄基丙酮)钯和双(三苯基膦)氯化钯(pd(pph3)2cl2)。在一个优选实施方式中,钯催化剂是乙酸钯。在一个优选实施方式中,钯催化剂是双(三苯基膦)氯化钯。
[0102]
在一个实施方式中,步骤a)的烷氧羰基化反应是在有机磷配体的存在下进行的。在一个实施方式中,有机磷配体选自:三苯基膦(pph3)、三叔丁基膦(ptbu3)、4,5
‑
双(二苯基膦)
‑
9,9
‑
二甲基氧杂蒽(xantphos)、2,2'
‑
双(二苯基膦)
‑
1,1'
‑
联萘(binap)、1,3
‑
双(二苯基膦)丙烷(dppp)和二
‑
(1
‑
金刚烷基)正丁基膦在一个优选实施方式中,有机磷配体是4,5
‑
双(二苯基膦)
‑
9,9
‑
二甲基氧杂蒽(xantphos)。在另一个实施方式中,有机磷配体是三苯基膦(pph3)。
[0103]
替代地,根据钯催化剂,步骤a)的烷氧羰基化反应在没有有机磷配体的情况下进行。例如,当使用双(三苯基膦)氯化钯(pd(pph3)2cl2)作为钯催化剂时,情况就是如此。
[0104]
在一个实施方式中,步骤a)的烷氧羰基化反应是在碱的存在下进行的。在一个实施方式中,碱选自乙酸钠(naoac)、n,n
‑
二异丙基乙胺(dipea)、二甲基吡啶(lutidine)、n
‑
甲基吗啉(nmm)、三丁胺、三乙胺(tea)及其混合物。在一个优选实施方式中,碱是乙酸钠。在一个实施方式中,碱在干燥形式下使用。
[0105]
在一个实施方式中,步骤a)在适于获得所需r2部分的溶剂中进行。在一个实施方式中,步骤a)在醇中进行,优选在甲醇或乙醇中进行。在一个实施方式中,步骤a)在醇溶剂和甲基叔丁基醚(mtbe)的混合物中进行,例如mtbe与甲醇或乙醇的混合物。在一个实施方式中,当r2是乙基时,步骤a)在乙醇中或在乙醇与mtbe的混合物中进行。溶剂可以是干燥的或非干燥的。在一个实施方式中,使用2
‑
30体积、优选5
‑
10体积、更优选约10体积的溶剂。
[0106]
在一个实施方式中,所使用的式(iii)化合物的浓度范围为0.01m
‑
1m,优选0.1m
‑
0.5m,更优选0.2m
‑
0.4m。
[0107]
在一个实施方式中,相对于化合物(iii),碱(例如乙酸钠)的摩尔当量数为1
‑
3,优选1.1
‑
2、更优选1.1
‑
1.5、更优选约1.3当量。
[0108]
在一个实施方式中,相对于化合物(iii),钯催化剂(例如乙酸钯)的摩尔当量数为0.003
‑
0.1,优选0.005
‑
0.05、更优选0.005
‑
0.01、更优选约0.005当量。
[0109]
在一个实施方式中,相对于化合物(iii),有机磷配体(例如xantphos)的摩尔当量数为0.003
‑
0.2,优选0.005
‑
0.15、更优选0.005
‑
0.05、更优选约0.005当量。
[0110]
在一个实施方式中,步骤a)在50℃
‑
150℃、优选63℃
‑
67℃、更优选约65℃的温度下进行。替代地,步骤a)可以在90℃
‑
120℃、优选100℃
‑
110℃的温度下进行。
[0111]
在一个实施方式中,步骤a)以至少3小时、优选10小时
‑
48小时,优选15小时
‑
40小时的时长进行。在一个实施方式中,步骤a)进行约19小时。在一个实施方式中,步骤a)进行约30小时。在一个实施方式中,步骤a)进行约37小时。反应持续时间与反应中涉及的量的规模相匹配。
[0112]
在一个实施方式中,完成后,所得产物用二氯甲烷、叔丁基甲醚或甲基环己烷萃取,优选用甲基环己烷萃取。
[0113]
在一个实施方式中,所得产物(ii)可以通过蒸馏纯化。
[0114]
在一个实施方式中,步骤a)的烷氧羰基化反应是在一氧化碳、钯催化剂、有机磷配
体、碱和醇溶剂的存在下进行的。在一个实施方式中,钯催化剂是乙酸钯,有机磷配体是4,5
‑
双(二苯基膦)
‑
9,9
‑
二甲基氧杂蒽(xantphos),碱是乙酸钠,溶剂是乙醇或mtbe与乙醇的混合物。在另一个实施方式中,钯催化剂是乙酸钯,有机磷配体是三苯基膦(pph3),碱是三乙胺,溶剂是乙醇或mtbe与乙醇的混合物。
[0115]
在另一个实施方式中,步骤a)的烷氧羰基化反应是在一氧化碳、钯催化剂、碱和醇溶剂的存在下进行的。在一个实施方式中,钯催化剂是双(三苯基膦)氯化钯(pd(pph3)2cl2),碱是三乙胺,溶剂是乙醇或mtbe与乙醇的混合物。
[0116]
fe(co)5烷氧羰基化反应
[0117]
在一个实施方式中,步骤a)的烷氧羰基化反应是在fe(co)5的存在下进行的。
[0118]
在一个实施方式中,步骤a)的烷氧羰基化反应是在一氧化碳、钯催化剂、有机磷配体、碱和fe(co)5的存在下在醇溶剂中进行的。上述关于烷氧羰基化条件和特别是关于这些组分的实施方式在用fe(co)5时也适用。
[0119]
在一个实施方式中,相对于化合物(iii),fe(co)5的摩尔当量数为0.01
‑
0.5,优选0.05
‑
0.2、更优选约0.01当量。
[0120]
通过锂交换的烷氧羰基化
[0121]
在一个实施方式中,步骤a)的烷氧羰基化反应是通过锂交换进行的。
[0122]
在一个实施方式中,步骤a)的烷氧羰基化反应是通过首先使式(iii)化合物与有机锂试剂接触、之后通过加入氯甲酸酯或氰基甲酸酯来进行的,以获得式(ii)化合物。
[0123]
在一个实施方式中,有机锂试剂选自正己基锂和正丁基锂,优选地,有机锂试剂为正己基锂。
[0124]
在一个实施方式中,氯甲酸酯选自:氯甲酸烷基酯和氯甲酸芳烷基酯,优选地,氯甲酸烷基酯选自氯甲酸乙酯、氯甲酸甲酯和氯甲酸正丁酯,氯甲酸芳烷基酯例如为氯甲酸苄酯,更优选地,氯甲酸酯是烷基氯甲酸酯,例如氯甲酸乙酯。
[0125]
在一个实施方式中,氰基甲酸酯选自:烷基氰基甲酸酯和芳烷基氰基甲酸酯,优选地,烷基氰基甲酸酯选自氰基甲酸乙酯、氰基甲酸甲酯和氰基甲酸叔丁酯,芳烷基氰基甲酸酯例如为氰基甲酸苄酯,更优选地,氰基甲酸酯是烷基氰基甲酸酯,如氰基甲酸乙酯。
[0126]
在一个实施方式中,相对于化合物(iii),有机锂试剂(优选己基锂)的摩尔当量数为1
‑
2,优选1
‑
1.5、更优选约1.1当量。
[0127]
在一个实施方式中,相对于化合物(iii)而言,氯甲酸酯(优选氯甲酸乙酯)的摩尔当量数为1
‑
10,优选4
‑
8、更优选约6当量。
[0128]
在一个实施方式中,锂交换是在惰性气氛下进行的。
[0129]
在一个实施方式中,锂交换是在干燥溶剂中进行的,例如在甲基四氢呋喃(methf)、四氢呋喃(thf)、叔丁基甲醚(tbme)、正己烷及其混合物中进行,优选在甲基四氢呋喃中进行。
[0130]
在一个实施方式中,锂交换在低于0℃的温度下进行,优选在约
‑
65℃下进行。
[0131]
在一个实施方式中,所得式(ii)化合物可以通过蒸馏纯化。
[0132]
步骤b)
‑
酰基肼的形成(步骤b)
[0133]
在步骤b)中,通过使式(ii)化合物与肼、优选一水合肼反应来形成式(i)化合物。
[0134]
在一个实施方式中,相对于式(ii)化合物,肼的摩尔当量数为1
‑
2,优选1.1
‑
1.2、
更优选约1.14当量。
[0135]
在一个实施方式中,步骤b)是在选自甲醇、乙醇和异丙醇的溶剂中进行的。在一个优选实施方式中,溶剂是异丙醇。在另一个优选实施方式中,溶剂是乙醇。
[0136]
在一个实施方式中,步骤b)是在低于10℃、优选0℃
‑
5℃的温度下进行的。
[0137]
在一个实施方式中,步骤b)以至少30分钟、优选至少1小时、优选1
‑
48小时、更优选1
‑
24小时的时长进行。
[0138]
在一个实施方式中,完成后,以固体形式回收式(i)化合物,可以用溶剂如甲醇或异丙醇对其进行洗涤。
[0139]
sandmeyer反应(预步骤)
[0140]
在一个实施方式中,本发明的方法包括对式(iv)化合物进行sandmeyer反应的预步骤,从而获得式(iii)化合物。例如,当x为br时,可以使用goerdeler等人的chem.ber.,1956,89,1534
‑
1540或us2007/0078155报道的方法获得化合物(iii
‑1‑
a)。
[0141]
在一个实施方式中,通过sandmeyer反应由式(iv)化合物获得式(iii)的卤化物化合物:
[0142][0143]
或其盐,其中r1代表甲基或甲基
‑
d3。
[0144]
在一个实施方式中,sandmeyer反应是通过首先在原位形成的亚硝酸的存在下由胺形成重氮盐、之后用卤化物阴离子作为亲核试剂将其取代来进行的。
[0145]
在一个实施方式中,通过sandmeyer反应的溴化是在亚硝酸钠(nano2)和溴化氢(hbr)的存在下进行的,优选在水性介质中进行。在这种情况下,在式(iii)化合物中,x是溴。
[0146]
在一个实施方式中,相对于化合物(iv),溴化氢的摩尔当量数为1
‑
5,优选2
‑
4、更优选约3当量。
[0147]
在一个实施方式中,相对于化合物(iv),亚硝酸钠的摩尔当量数为1.15至4,优选1.5
‑
2、更优选约1.5当量。
[0148]
在一个实施方式中,sandmeyer反应在室温至60℃、优选35℃
‑
50℃、更优选约40℃
‑
约45℃的温度下进行。
[0149]
在一个实施方式中,sandmeyer反应以至少30分钟、优选30分钟
‑
22小时、优选30分钟
‑
2小时、更优选约1小时的时长进行。
[0150]
在一个实施方式中,sandmeyer反应是在1摩尔当量的式(iv)的胺化合物、3摩尔当量的溴化氢和1.5摩尔当量的亚硝酸钠的存在下在水中进行的。优选地,反应在约40℃的温度下进行。优选地,反应在5体积的水中进行。
[0151]
在一个实施方式中,完成后,通过用二氯甲烷萃取来回收产物。优选地,使用naoh溶液中和有机相。
[0152]
在另一个实施方式中,通过sandmeyer反应的溴化是在叔丁基亚硝酸酯(tbuono)和cubr2的存在下进行的。在这种情况下,在式(iii)化合物中,x是溴。
[0153]
在一个实施方式中,相对于化合物(iv),cubr2的摩尔当量数为1
‑
3,优选1
‑
1.1、更
优选约1.03当量。
[0154]
在一个实施方式中,相对于化合物(iv),叔丁基亚硝酸酯的摩尔当量数为1
‑
3,优选1
‑
2、更优选约1.5当量。
[0155]
在一个实施方式中,sandmeyer反应在0℃
‑
40℃、优选0℃至室温的温度下进行。
[0156]
在一个实施方式中,sandmeyer反应以至少30分钟、优选30分钟
‑
10小时、优选30分钟
‑
5小时、更优选2小时
‑
3小时的时长进行。
[0157]
在一个实施方式中,sandmeyer反应在1摩尔当量的式(iv)的胺化合物、1.03摩尔当量的cubr2和1.5摩尔当量的叔丁基亚硝酸酯的存在下在乙腈中进行。优选地,反应首先在约0
‑
5℃的温度下进行,之后在室温下进行。
[0158]
在一个实施方式中,完成后,通过用叔丁基甲醚萃取回收产物。
[0159]
在另一个实施方式中,通过sandmeyer反应的碘化是在叔丁基亚硝酸酯(tbuono)和碘的存在下进行的。在这种情况下,在式(iii)化合物中,x是碘。优选地,该反应在诸如乙腈等溶剂中进行。
[0160]
在一个实施方式中,相对于化合物(iv),碘的摩尔当量数为1
‑
3,优选1
‑
2、更优选约1当量。
[0161]
在一个实施方式中,相对于化合物(iv),叔丁基亚硝酸酯的摩尔当量数为1
‑
5,优选3
‑
5、更优选约4当量。
[0162]
在一个实施方式中,sandmeyer反应在从室温到回流的温度范围内进行。
[0163]
在一个实施方式中,sandmeyer反应以至少30分钟、优选30分钟
‑
5小时、优选30分钟
‑
2小时、更优选约1小时的时长进行。
[0164]
在一个实施方式中,sandmeyer反应在1摩尔当量的式(iv)的胺化合物、1摩尔当量的碘和4摩尔当量的叔丁基亚硝酸酯的存在下进行。
[0165]
在一个实施方式中,完成后,将过量的碘淬灭,优选通过加入na2so3来淬灭。之后,获得的化合物可以用甲基叔丁基醚萃取。
[0166]
在另一个实施方式中,通过sandmeyer反应的碘化是在亚硝酸钠(nano2)、碘化钾(ki)和对甲苯磺酸(tsoh)的存在下进行的。在这种情况下,在式(iii)化合物中,x是碘。
[0167]
在一个实施方式中,相对于化合物(iv),碘化钾的摩尔当量数为1
‑
4,优选2
‑
3、更优选约2.6当量。
[0168]
在一个实施方式中,相对于化合物(iv),亚硝酸钠的摩尔当量数为1
‑
3,优选1.5
‑
2.5、更优选约2当量。
[0169]
在一个实施方式中,相对于化合物(iv),对甲苯磺酸的摩尔当量数为1
‑
6,优选3
‑
4、更优选约3.5当量。
[0170]
在一个实施方式中,sandmeyer反应优选在室温下进行。
[0171]
在一个实施方式中,sandmeyer反应以至少1小时、优选1小时
‑
24小时、优选约12小时的时长进行。
[0172]
在一个实施方式中,sandmeyer反应在1摩尔当量的式(iv)的胺化合物、2.6摩尔当量的碘化钾、约2摩尔当量的亚硝酸钠和3.5摩尔当量的对甲苯磺酸的存在下进行。
[0173]
氘代化合物(iv
‑
2)的合成
[0174]
非氘代中间体(iv
‑
1),即式(iv)化合物,其中r1是甲基,也称为amtd,是市售的或
可以通过本领域技术人员已知的方法获得。
[0175]
对于相应的氘代中间体(iv
‑
2),即式(iv)化合物,其中r1是甲基
‑
d3,从非氘代的合成路线无法预测可以达到何种同位素纯度。本文提供了一种合成中间体(iv
‑
2)的方法,其能够实现非常高的同位素纯度,即总同位素纯度大于90%,更优选大于95%。
[0176]
d3
‑
乙脒路线
[0177]
因此,本发明还涉及3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
胺(iv
‑
2)或其盐的制备方法:
[0178][0179]
其包括以下步骤:
[0180]
a)在hcl的存在下使d3
‑
乙腈与乙醇反应,以形成式(vi
‑
2)的pinner盐:
[0181][0182]
b)使pinner盐(vi
‑
2)与氨反应,以形成d3
‑
乙脒(v
‑
2)或其盐:
[0183][0184]
以及
[0185]
c1)使d3
‑
乙脒(v
‑
2)与溴、硫氰酸盐和甲醇钠反应以获得式(iv
‑
2)化合物;或
[0186]
c2)使d3
‑
乙脒(v
‑
2)首先与次氯酸钠(naocl)反应,之后与硫氰酸盐反应,以获得式(iv
‑
2)化合物。
[0187]
在该方法中,步骤a)通过pinner反应形成pinner盐。“pinner反应”是指腈(本文中为d3
‑
乙腈)与醇(本文中为乙醇)的酸催化反应而形成的亚氨基醚盐,也称为pinner盐。pinner盐本身具有反应性,并且例如与氨经过额外的亲核加成形成脒,如步骤b)所示。
[0188]
最后一步可以通过“溴路线”(步骤c1)或通过“次氯酸盐路线”(步骤c2)进行,两者都提供了令人满意的结果。
[0189]
如上所述,最终化合物或本发明的方法中使用的中间体可以是盐的形式,包括酸加成盐和碱盐。
[0190]
步骤a)
‑
pinner盐的形成
[0191]
在步骤a)中,通过使d3
‑
乙腈与乙醇在hcl的存在下反应而获得式(vi
‑
2)的pinner盐。
[0192]
在一个实施方式中,在步骤a)中,通过使hcl气体鼓泡通过包含乙醇(优选无水乙醇)和d3
‑
乙腈的混合物来形成pinner盐(vi
‑
2)。
[0193]
在一个实施方式中,hcl鼓泡在低于15℃、优选低于10℃的温度下进行。
[0194]
在一个实施方式中,以至少10小时、优选至少8小时、更优选约6小时的时长在混合物中鼓泡hcl气体。
[0195]
在一个实施方式中,在hcl鼓泡结束后,在室温下、优选在20℃
‑
25℃的温度范围内搅拌反应混合物。
[0196]
在一个实施方式中,在hcl鼓泡结束后,以至少10小时、优选10小时
‑
24小时、优选
15小时
‑
20小时、更优选约16.5小时的时长搅拌反应混合物。
[0197]
在一个实施方式中,完成后,用叔丁基甲醚(tbme)处理反应混合物,优选在0℃
‑
5℃的温度范围内处理,从而以固体形式分离pinner盐(vi
‑
2)。
[0198]
步骤b)
‑
氘代乙脒的形成
[0199]
在步骤b)中,通过使pinner盐(vi
‑
2)与氨(nh3)反应来形成氘代乙脒(v
‑
2)。
[0200]
在一个实施方式中,步骤b)中使用的溶剂是醇,例如乙醇或甲醇,优选乙醇,更优选无水乙醇。
[0201]
在一个实施方式中,步骤b)在低于10℃的温度下进行,优选在0℃
‑
5℃的温度范围内进行。
[0202]
在一个实施方式中,氨(nh3)作为气体使用并被直接吸收在反应混合物中。优选地,相对于pinner盐(vi
‑
2),使用3
‑
5摩尔当量、优选4.1摩尔当量的氨。
[0203]
在一个实施方式中,步骤b)的反应以至少1小时、优选1
‑
6小时,优选2
‑
4小时、更优选约3小时的时长进行。
[0204]
在一个实施方式中,完成后,将反应混合物蒸发并用甲基环己烷处理,从而以固体形式回收d3
‑
乙脒(v
‑
2)。
[0205]
有利地,获得的d3
‑
乙脒(v
‑
2)的同位素纯度为至少90%,优选至少95%。
[0206]
步骤c1)
‑
通过“溴路线”环化形成噻二唑环
[0207]
获得式(iv
‑
2)化合物的最后一步可以通过“溴路线”(步骤c1)进行。
[0208]
在步骤c1)中,通过使d3
‑
乙脒(v
‑
2)与溴、硫氰酸盐和甲醇钠反应获得式(iv
‑
2)化合物。
[0209]
在一个实施方式中,硫氰酸盐优选以硫氰酸钾的形式使用。
[0210]
在一个实施方式中,甲醇钠可以通过在甲醇中溶解钠来获得。
[0211]
在一个实施方式中,相对于d3
‑
乙脒(v
‑
2),硫氰酸盐的摩尔当量数为1至2.6,优选1.1当量。
[0212]
在一个实施方式中,相对于d3
‑
乙脒(v
‑
2),甲醇钠(naome)的摩尔当量数为2
‑
5,优选3
‑
3.5、更优选约3.1当量。
[0213]
在一个实施方式中,相对于d3
‑
乙脒(v
‑
2),溴(br2)的摩尔当量数为1
‑
3,优选1
‑
2、更优选约1.5当量。
[0214]
在一个实施方式中,步骤c1)中使用的溶剂是醇,优选甲醇。优选地,溶剂是干燥的,优选干燥甲醇。
[0215]
在一个实施方式中,步骤c1)在低于10℃的温度下进行,优选在0℃
‑
5℃的温度范围内进行。
[0216]
在一个实施方式中,步骤c1)的反应以至少1小时、优选1
‑
6小时、优选1
‑
3小时、更优选约2小时的时长进行。
[0217]
在一个实施方式中,用乙酸乙酯进行萃取并以固体形式获得产物。
[0218]
有利地,获得的噻二唑产物(iv
‑
2)的同位素纯度为至少90%。
[0219]
步骤c2)
‑
通过“次氯酸盐路线”环化形成噻二唑环
[0220]
替代地,获得式(iv
‑
2)化合物的最后一步可以通过“次氯酸盐路线”(步骤c2)进行。
[0221]
在步骤c2)中,通过首先使d3
‑
乙脒(v
‑
2)与次氯酸钠(naocl)反应,形成中间体(viii
‑
2)或其盐:
[0222][0223]
之后使形成的中间体(viii
‑
2)与硫氰酸盐反应,来获得式(iv
‑
2)化合物。
[0224]
在一个实施方式中,次氯酸钠(naocl)以水溶液的形式使用,并且可以通过在水中溶解naocl五水合物来获得。
[0225]
在一个实施方式中,相对于d3
‑
乙脒(v
‑
2),次氯酸钠(naocl)的摩尔当量数为1
‑
5,优选为1.5
‑
2。
[0226]
在一个实施方式中,与次氯酸钠的反应所使用的溶剂是水,优选去离子水。
[0227]
在一个实施方式中,与次氯酸钠的反应在低于10℃的温度下进行,优选在0℃
‑
5℃的温度范围内进行。
[0228]
在一个实施方式中,与次氯酸钠的反应以至少30分钟、优选1
‑
6小时、优选1
‑
3小时、更优选约2小时的时长进行。
[0229]
在一个实施方式中,所得中间体(viii
‑
2)用乙酸乙酯萃取。
[0230]
在一个实施方式中,硫氰酸盐优选以硫氰酸钠的形式使用。
[0231]
在一个实施方式中,相对于中间体(viii
‑
2),硫氰酸盐的摩尔当量数为1
‑
2,优选1
‑
1.5、更优选约1.1当量。
[0232]
在一个实施方式中,中间体(viii
‑
2)与硫氰酸盐反应所使用的溶剂是醇,优选甲醇。优选地,溶剂是干燥的,优选干燥甲醇。
[0233]
在一个实施方式中,中间体(viii
‑
2)与硫氰酸盐的反应在低于10℃的温度下进行,优选在0℃
‑
5℃的温度范围内进行。
[0234]
在一个实施方式中,中间体(viii
‑
2)与硫氰酸盐的反应以至少30分钟、优选1
‑
6小时、优选1
‑
3小时,更优选约1.5小时的时长进行。
[0235]
在一个实施方式中,用乙酸乙酯进行萃取并以固体形式获得产物。
[0236]
有利地,获得的噻二唑产物(iv
‑
2)的同位素纯度为至少95%。
[0237]
d3
‑
羟基乙脒路线
[0238]
本发明还涉及另一种制备3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
胺(iv
‑
2)或其盐的方法:
[0239][0240]
其包括以下步骤:
[0241]
a)使d3
‑
乙腈与羟胺(h2n
‑
oh)反应,以形成d3
‑
羟基乙脒(ix
‑
2)或其盐:
[0242][0243]
b)对d3
‑
羟基乙脒(ix
‑
2)进行甲苯磺酰基化,以形成中间体(x
‑
2)或其盐:
[0244][0245]
以及
[0246]
c)使中间体(x
‑
2)与硫氰酸盐反应,以获得式(iv
‑
2)化合物。
[0247]
步骤a)
‑
氘代羟基乙脒的形成
[0248]
在步骤a)中,使d3
‑
乙腈与羟胺(h2n
‑
oh)反应获得d3
‑
羟基乙脒(ix
‑
2)。
[0249]
在一个实施方式中,羟胺可以以盐酸羟胺或羟胺的水溶液的形式使用。
[0250]
在一个实施方式中,相对于d3
‑
乙腈,羟胺的摩尔当量数为1
‑
4,优选2
‑
2.5、更优选约2.2当量。
[0251]
在一个实施方式中,步骤a)中使用的溶剂是醇,优选乙醇。
[0252]
在一个实施方式中,步骤a)在回流温度下进行。
[0253]
在一个实施方式中,步骤a)的反应以至少1小时、优选1
‑
24小时、优选约18小时的时长进行。
[0254]
有利地,同位素纯度在步骤a)期间得以保留。
[0255]
步骤b)
‑
氘代羟基乙脒的甲苯磺酰基化
[0256]
在步骤b)中,在d3
‑
羟基乙脒(ix
‑
2)上进行甲苯磺酰基化反应,以获得相应的甲苯磺酰基化中间体(x
‑
2)。甲苯磺酰基化可以在对甲苯磺酰氯和碱的存在下进行。
[0257]
在一个实施方式中,碱选自:三乙胺(tea)、乙酸钠(naoac)、n,n
‑
二异丙基乙胺(dipea)、n
‑
甲基吗啉(nmm)及其混合物。在一个优选实施方式中,碱是三乙胺。
[0258]
在一个实施方式中,相对于d3
‑
羟基乙脒(ix
‑
2),对甲苯磺酰氯的摩尔当量数为0.8
‑
1.5,优选0.9
‑
1.1、更优选约0.9当量。
[0259]
在一个实施方式中,相对于d3
‑
羟基乙脒(ix
‑
2),碱(例如三乙胺)的摩尔当量数为1
‑
2,优选1
‑
1.5、更优选约1.3当量。
[0260]
在一个实施方式中,步骤b)中使用的溶剂是四氢呋喃,优选无水四氢呋喃。
[0261]
在一个实施方式中,对甲苯磺酰氯的加入在低于10℃的温度下进行,优选在0℃
‑
5℃的温度范围内进行。在完成对甲苯磺酰氯的加入后,反应优选在室温下进行。
[0262]
在一个实施方式中,步骤b)的反应以至少30分钟、优选30分钟
‑
3小时、优选约1小时的时长进行。
[0263]
在一个实施方式中,用乙酸乙酯萃取产物。
[0264]
有利地,同位素纯度在步骤b)期间得以保留。
[0265]
步骤c)
‑
环化形成噻二唑环
[0266]
在步骤c)中,使d3
‑
甲苯磺酰氧基乙脒(x
‑
2)与硫氰酸盐反应获得式(iv
‑
2)化合物。
[0267]
在一个实施方式中,硫氰酸盐优选以硫氰酸钾的形式使用。
[0268]
在一个实施方式中,步骤c)可以任选地在碱的存在下进行。当使用碱时,其可以选自:n,n
‑
二异丙基乙胺(dipea)、乙酸钠(naoac)、三乙胺(tea)、n
‑
甲基吗啉(nmm)及其混合物。
[0269]
在一个实施方式中,相对于d3
‑
甲苯磺酰氧基乙脒(x
‑
2),硫氰酸盐的摩尔当量数为1
‑
5,优选2至4、更优选约3当量。
[0270]
在一个实施方式中,相对于d3
‑
甲苯磺酰氧基乙脒(x
‑
2),碱(例如dipea)的摩尔当量数为0
‑
2,优选1
‑
2、更优选1
‑
1.5、仍更优选约1.1当量。
[0271]
在一个实施方式中,步骤c)中使用的溶剂选自:醇(例如甲醇)、二甲基甲酰胺(dmf)、乙腈、四氢呋喃(thf)、二甲基亚砜(dmso)及其混合物。在一个实施方式中,步骤c)中使用的溶剂是甲醇。
[0272]
在一个实施方式中,步骤c)在20℃
‑
50℃的温度范围内进行,优选在约40℃的温度下执行。
[0273]
在一个实施方式中,步骤c)的反应以至少1小时、优选2
‑
20小时的时长进行。
[0274]
有利地,获得的噻二唑产物(iv
‑
2)的同位素纯度为至少95%。
[0275]
实施例
[0276]
本发明通过以下实施例进一步说明。实施例部分所述的反应方案以举例的方式说明了不同的可能方法。
[0277]
材料和方法
[0278]
所有报告的温度均以摄氏度(℃)表示;除非另有说明,否则所有反应均在室温(rt)下进行。
[0279]
分析方法:
[0280]
采用分析薄层色谱(tlc)对反应进行监测,建立快速色谱条件,验证中间体或最终产物的纯度。所使用的tlc板为merck tlc铝片硅胶60f
254
。在室温下用紫外光照射(波长=254nm)或加热到160℃用kmno4显示剂显示tlc板。kmno
4 tlc显示剂通过将3g高锰酸钾、20g碳酸钠溶于300ml蒸馏水中制得。
[0281]
在bruker avance 500mhz上记录1h和
13
c nmr谱。化学位移以百万分之一(ppm,δ单位)表示。耦合常数用赫兹(hz)表示。分裂模式体现出表观多重性,并且表示为s(单重态)、d(双重态)、t(三重态)、q(四重态)、h(六重态)、m(多重态)或br(宽峰)。
[0282]
gc测定通过以下方式进行:(条件a)在varian 3900上用火焰离子化监测器(fid)在260℃下进行,所用柱为rtx
‑
1301,30m
×
0.32mm
×
0.5μm,喷射器温度为200℃,分流比为50,注射量为1μl,采用以下温度程序:烘箱温度设置为60℃下4分钟,以20℃/分钟的速率线性增加到260℃,并且在260℃下保持10分钟;或(条件b)在shimadzu gc
‑
2010上用火焰离子化监测器(fid)在260℃下进行,喷射器温度为200℃,分流比为50,注射量为1μl,所用柱为rxi
‑
17sil ms,30m
×
0.32mm
×
0.25μm,采用以下温度程序:烘箱温度设置为60℃下5分钟,以20℃/分钟的速率线性增加到300℃,并且在300℃下保持8分钟;或(条件c)在varian 3900上用火焰离子化监测器(fid)在270℃下进行,所用柱为db
‑
624,60m
×
0.32mm
×
3μm,喷射器温度为200℃,分流比为50,注射量为1μl,采用以下温度程序:烘箱温度设置为80℃下4分钟,以20℃/分钟的速率线性增加到260℃,并且在260℃下保持11分钟。
[0283]
gc
‑
ms分析在shimadzu gcms
‑
qp2010上用电子电离监测器(ei)在250℃下进行。所用柱为phenomenex xb
‑
5ms,30m
×
0.25mm
×
0.25μm。柱流量为1.50ml/分钟。喷射器温度为250℃,分流比为50。注射量为1μl。采用以下温度程序:在最初的40℃下保持5分钟,之后以20℃/分钟升温到250℃,之后在250℃下保持5分钟。
[0284]
hplc谱在装有uv监测器的thermo science ultimate 3000hplc装置上获得。
[0285]
条件a:所用柱为phenomenex
‑
kinetex evo c18 50
×
4.6mm
×
2.6μl。洗脱液为溶液a(10mm hco2nh4的h2o溶液)和溶液b(mecn)的混合物。在1.0ml分钟
‑1的流速下施加梯度:保持1%溶液b的初始条件3分钟,在7分钟内线性增加到90%溶液b,在2分钟内保持90%,在1.0分钟内回到初始条件并保持5分钟。
[0286]
条件b:所用柱为purospher star rp18e 55mm
×
4.6mm
×
3.0μm。洗脱液为溶液a(20mm hco2nh4的h2o溶液)和溶液b(mecn)的混合物。在1.0ml分钟
‑1的流速下施加梯度:保持2%溶液b的初始条件1分钟,在8分钟内线性增加到90%溶液b,在2分钟内保持90%,在1.0分钟内回到初始条件并保持3分钟。
[0287]
hplc
‑
ms谱在shimadzu lcms
‑
2020上用210nm uv监测器和电喷雾电离(esi)获得。在选定的离子监测模式下,通过比较各同位素的峰面积来确定同位素纯度。
[0288]
条件a:所用柱为sequant zic
‑
hilic 150
×
4.6mm
×
5μm。洗脱液为25%溶液a(20mm nh4ac的h2o溶液)和75%溶液b(mecn)的混合物,流速为1ml分钟
‑1。
[0289]
条件b:所用柱为ymc
‑
triart c18 100
×
3.0mm
×
3μm。洗脱液为溶液a(0.1%hco2h的h2o溶液)和溶液b(mecn)的混合物。在0.5ml分钟
‑1的流速下施加梯度:保持0%溶液b的初始条件12分钟,在5分钟内线性增加到90%溶液b,在6分钟内保持90%,在0.1分钟内回到初始条件并保持7分钟。
[0290]
条件c:所用柱为phenomenex
‑
kinetex evo c18 100
×
2.1mm
×
2.6μm。洗脱液为溶液a(0.1%hco2h的h2o溶液)和溶液b(mecn)的混合物。在0.5ml分钟
‑1的流速下施加梯度:保持2%溶液b的初始条件1分钟,在9分钟内线性增加到90%溶液b,在3分钟内保持90%,在0.1分钟内回到初始条件并保持5分钟。
[0291]
条件d:所用柱为ymc
‑
triart c18 100
×
3.0mm
×
3μm。洗脱液为溶液a(0.1%hco2h的h2o溶液)和溶液b(mecn)的混合物。在0.5ml分钟
‑1的流速下施加梯度:保持90%溶液b的初始条件23分钟,在0.1分钟内线性下降到1%溶液b并保持7分钟。
[0292]
替代地,hplc
‑
ms谱在agilent lcms上用电喷雾电离(esi)获得。agilent装置包括自动取样器1100、二元泵1100、紫外多波长监测器1100和6100单四联质谱仪。条件d:所用柱为sunfire 3.5μm,c18,3.0
×
50mm。洗脱液为溶液a(0.1%tfa的h2o溶液)和溶液b(0.1%tfa的mecn溶液)的混合物。在1.3ml分钟
‑1的流速下施加梯度(用于分析最终化合物和中间体):保持5%溶液b的初始条件0.2分钟,在6分钟内线性增加到95%溶液b,在1.75分钟内保持95%,在0.25分钟内返回初始条件并保持2.0分钟。
[0293]
氯离子含量用mettler toledo dl50滴定仪或等效的滴定仪、dm
‑
141或等效的电极、分析天平、标准实验室玻璃器皿获得。所用化学试剂为,滴定剂:0.1mol/l硝酸银(0.1mol/l硝酸银的制备:称取约17g硝酸银(agno3),放入1000ml容量瓶中,溶解并用去离子水稀释至刻度)。测定溶液的因子。样品的制备:在分析天平上称取30
‑
500mg固体样品,放入容器中,作为卤化物含量的函数。用溶剂(去离子水、2
‑
丙醇等)稀释至60ml。结果评价:
[0294][0295]
[0296]
式中,q1:当量点的滴定剂(mmol),m
x
:摩尔质量(mg/mmol),m:样品重量(mg),v:样品体积(μl)
[0297]
卤化物氯化物m
x
(mg/mmol)35.45
[0298]
基于氯离子含量的测定通过氯化物测定来计算:
[0299][0300]
其中,97.56g/mol为化合物(v
‑
2)的分子量,53.45g/mol为nh4cl的分子量,m
产物
为分离的hcl盐的质量,w
cl
‑
为滴定测得的氯离子的重量百分比。
[0301]
反应物
[0302]
除另有说明外,溶剂、试剂和原料均按从商业供应商处购买和使用。
[0303]
使用以下缩写:
[0304]
amtd:5
‑
氨基
‑3‑
甲基
‑
1,2,4
‑
噻二唑,
[0305]
cca.:大约,
[0306]
dcm:二氯甲烷,
[0307]
eq:当量,
[0308]
etoac:乙酸乙酯,
[0309]
etoh:乙醇,
[0310]
g:克,
[0311]
gc:气相色谱,
[0312]
hex:正己烷,
[0313]
hplc:高效液相色谱,
[0314]
ipa:异丙醇,
[0315]
l:升,
[0316]
lcms:液相色谱质谱联用,
[0317]
mecn:乙腈,
[0318]
meoh:甲醇,
[0319]
ml:毫升,
[0320]
mol:摩尔,
[0321]
mmol:毫摩尔,
[0322]
min:分钟,
[0323]
ms:质谱法,
[0324]
nmm:n
‑
甲基吗啉
[0325]
mw:分子量,
[0326]
nmr:核磁共振,
[0327]
rt:室温,
[0328]
tbme:叔丁基甲醚,
[0329]
tea:三乙胺,
[0330]
thf:四氢呋喃,
[0331]
tlc:薄层色谱法,
[0332]
ts:甲苯磺酰基(即对甲苯磺酰基)
[0333]
vol.:体积。
[0334]
本技术中公开的所有化合物使用从cambridgesoft(剑桥,马萨诸塞州,美国)购买的chemdraw ultra命名。
[0335]
实施例1a:通过乙脒路线合成氘代d3
‑
amtd(iv
‑
2)
[0336][0337]
通过pinner盐(vi
‑
2)进行pinner反应获得d3
‑
乙脒(v
‑
2),之后通过“溴路线”(步骤c1)或“次氯酸盐路线”(步骤c2)环化,以形成化合物(iv
‑
2)的噻二唑环。
[0338]
步骤a:pinner盐的形成(vi
‑
2)
[0339]
在20
‑
25℃下,在装有两个进气口、温度计和磁力搅拌子的750ml三颈玻璃烧瓶中,装入74ml无水二醇和51.2g(1.161mol,1.0eq)d3
‑
乙腈(vii
‑
2),之后冷却到0
‑
5℃(通过冰盐混合物冷却)。将温度保持在10℃以下,使hcl气体鼓泡通过混合物,持续6小时。将反应混合物加热至20
‑
25℃并搅拌16.5小时(混合物变成难以搅拌的黏稠白色悬浮液)。向混合物中加入500ml tbme,之后混合物变成容易搅拌的白色悬浮液。在0
‑
5℃下搅拌1小时,之后过滤,用2
×
50ml冷tbme洗涤。过滤后的固体在40℃真空下干燥,获得104.3g白色固体pinner盐(vi
‑
2),其在下一步中使用而无需进一步纯化。
[0340]
步骤b:d3
‑
乙脒(v
‑
2)的形成
[0341]
在20
‑
25℃下,在装有两个进气口、温度计和磁力搅拌子的2l三颈玻璃烧瓶中,装入835ml(658.8克)无水乙醇,之后冷却至0
‑
5℃(通过冰盐混合物冷却)。在该温度下,吸收57.3g(3.37mol,4.1eq)nh3气体。之后,在0
‑
5℃下,加入104.3g(823.8mmol,1.0eq)pinner盐(vi
‑
2),搅拌3小时。用hplc对反应进行监测。在30℃下将反应混合物在旋转蒸发器上蒸发至干燥。向残留物中加入200ml甲基环己烷,并且在0
‑
5℃(通过冰浴冷却)下搅拌15分钟。将白色沉淀过滤,用25ml冷甲基环己烷洗涤,在45℃真空下干燥。获得77.1g(790.3mmol)白色固体d3
‑
乙脒盐酸盐(v
‑
2)。校正产率:89.3%。1h
‑
nmr(dmso
‑
d6):δ9.20(br,1h),8.75(br,2h),7.51(br,1h)。hplc
‑
ms(条件a)同位素纯度:95.8%。氯离子含量:38.46w/w%。基于氯离子含量的测定:93.1w/w%。
[0342]
步骤c1:通过溴路线合成d3
‑
amtd(iv
‑
2)
[0343]
在20
‑
25℃下,在装有两个滴液漏斗、温度计和磁力搅拌子的250ml四颈玻璃烧瓶中,装入5.8g(59.45mmol,1.0eq)d3
‑
乙脒盐酸盐(v
‑
2)(同位素纯度:96.5%,基于氯化物含
量的测定:64.7w/w%),6.35g(65.4mmol,1.1eq)kscn和11ml干燥meoh(用分子筛干燥)。在搅拌下将该混合物(悬浮液)冷却到10℃以下。将温度保持在0
‑
5℃下,在25分钟内同时向混合物中加入42.6ml(184.3mmol,3.1eq)25w/w%naome的meoh溶液(54.03g/mol)和4.6ml(14.26g,89.16mol,1.5eq)br2(纯净物,mw:159.81g/mol,d:3.119g/ml)溶液。加入后,混合物变为灰紫色,之后形成白色悬浮液。加入后,在0
‑
10℃下将混合物搅拌2小时,用tlc进行监测(洗脱液ch2cl2:meoh=5:1)。向混合物中逐渐加入20ml水,将温度在25℃以下。在环境温度下将混合物搅拌15分钟,之后在减压(80
‑
120毫巴,40℃)下蒸馏除去40ml meoh。水性残留物(约40ml)形成悬浮液,将其过滤。用3
×
40ml的etoac洗涤滤饼,获得6.95g白色固体副产物。将各份滤液用于萃取第一水性滤液。水性滤液的萃取:3
×
40ml etoac(滤饼的三次洗涤),再用3
×
40ml etoac进行萃取。用50ml饱和nacl溶液对合并的有机相进行洗涤。萃取物在40℃(100
‑
120毫巴)真空下浓缩。将形成的浆料(30ml)加热并回流30分钟,之后冷却至0
‑
5℃。在0
‑
5℃下放置过夜后,过滤悬浮液并用2
×
5ml冷etoac洗涤。所得产物(iv
‑
2)为灰白色固体,1.62g。校正产率:30%。同位素纯度:hplc
‑
ms(条件b)为93.4%,1h
‑
nmr为93%。滴定测定:85w/w%。1h
‑
nmr(dmso
‑
d6):δ7.77(br,2h),2.18(s,0.07h
‑
与cd2h信号有关)。
[0344]
步骤c2:通过次氯酸盐路线合成d3
‑
amtd(iv
‑
2)
[0345]
在20
‑
25℃下,在装有两个滴液漏斗、温度计和磁力搅拌子的500ml四颈玻璃烧瓶中,装入15.15g(132.5mmol,1.0eq)d3
‑
乙脒盐酸盐(v
‑
2)(同位素纯度:96.7%,基于氯化物含量的测定:85.7w/w%)和75ml(5体积)去离子水,之后冷却到0
‑
5℃。将温度保持在0
‑
5℃,在40分钟内将溶解在160ml(10.5体积)水中的26.95g(164mmol,1.24eq)五水合naocl加入到反应混合物中。反应用tlc(ch2cl2:meoh=5:1)进行监测。搅拌1小时后加入另一部分试剂:将温度保持在0
‑
5℃下,加入溶解在80ml(5.25体积)水中的13.48g(82mmol,0.62eq)五水合naocl。混合物在0
‑
5℃下搅拌1小时。之后,向水性混合物中加入85gnacl,之后用3
×
200ml etoac萃取,之后用na2so4干燥,过滤并真空蒸发(40℃,10毫巴)。中间体(viii
‑
2):10.0红色油状物。
[0346]
之后,在装有两个滴液漏斗、温度计和磁力搅拌子的500ml四颈玻璃烧瓶中,装入10.0g中间体(viii
‑
2)和100ml干燥meoh(用分子筛干燥),之后冷却至0
‑
5℃。在20分钟内将11.75g(145.5mmol,1.1eq)nascn小心地加入到该溶液中。将形成的悬浮液搅拌1.5小时,之后用60ml水淬灭。之后,将反应混合物过滤,之后在40℃和100
‑
200毫巴真空下蒸发掉meoh。将残留物(80ml)过滤,沉淀用3
×
100ml etoac洗涤。对每次洗涤的母液进行萃取,之后用100ml etoac。将有机相(4
×
100ml)合并并浓缩至27ml。将残留物回流15分钟,之后冷却至0
‑
5℃,静置1小时,过滤。过滤后的材料就是产物。粗产物:26%(4.1g)淡黄色固体。校正产率:12%(1.93g)。hplc(条件b):47w/w%。hplc
‑
ms(条件b)同位素纯度:96.8%。1h
‑
nmr(dmso
‑
d6):δ7.77(br,2h),3.46(br,1h),2.18(s,0.03h
‑
与cd2h信号有关)。
[0347]
实施例1b:通过羟基乙脒路线合成氘代d3
‑
amtd(iv
‑
2)
[0348][0349]
由氘代乙腈(vii
‑
2)获得d3
‑
羟基乙脒(ix
‑
2)。之后,在硫氰酸盐的存在下将相应
的甲苯磺酰基中间体(x
‑
2)环化,以形成化合物(iv
‑
2)的噻二唑环。
[0350]
步骤a:d3
‑
羟基乙脒(ix
‑
2)的形成
[0351]
在20
‑
25℃下,在装有回流冷凝器、温度计和磁力搅拌子的100ml玻璃容器中,装入5.5ml(4.65g,105.4mmol,1.0eq)d3
‑
乙腈(d:0.844g/ml)、33ml etoh和25.1ml(27.87g,422.3mmol,4.0eq)羟胺水溶液(50w/w%,mw:33g/mol,d:1.11g/ml)。将混合物加热至回流温度,在回流温度下搅拌5小时。在40℃真空下将溶剂从反应混合物中蒸发掉。所得产物(ix
‑
2)为白色固体。产率:84%(6.84g)。hplc
‑
ms(条件c)同位素纯度:98.5%。1h
‑
nmr(dmso
‑
d6):δ8.67(br,1h),5.33(br,2h)。
13
c
‑
nmr(dmso
‑
d6):δ149.9,16.3。
[0352]
步骤b:d3
‑
羟基乙脒甲苯磺酰基化形成化合物(x
‑
2)
[0353]
在20
‑
25℃下,在装有温度计和磁力搅拌子的500ml玻璃容器中,装入:6.3g(81.74mmol,1.0eq)d3
‑
羟基乙脒(ix
‑
2)和170ml(151g)干燥thf(通过分子筛干燥)。在环境温度下搅拌30分钟,之后在20
‑
25℃下加入14.8ml(10.75g,106.3mmol,1.3eq)tea。搅拌30分钟后,加入45ml干燥thf,但是仍有一些未溶解的物质残留在混合物中。用冰盐浴将混合物冷却至0
‑
5℃,之后在15分钟内分批向混合物中加入14.02g(73.57mmol,0.9eq)对甲苯磺酰氯。加入后,去除冷却浴,使得混合物升温到20
‑
25℃,并且在20
‑
25℃下搅拌一小时。混合物变成白色悬浮液。tlc监测:ch2cl2:meoh=95:5。完成后(对甲苯磺酰氯完全消耗),过滤沉淀(9.9g白色固体),用2
×
40ml thf洗涤。滤液在40℃真空下蒸发至干燥。向残留物中加入130ml(118g)etoac,有机相用1
×
65ml水和1
×
65ml饱和nacl溶液洗涤。水洗后,有机层用na2so4干燥,过滤,用2
×
20ml etoac洗涤。在40℃真空下将溶剂蒸发,获得无色油状物,其在静置后固化。产率:84%(15.9g,68.88mmol)。hplc
‑
ms(条件c)同位素纯度得以保留(98.5%)。
[0354]
步骤c:d3
‑
amtd(iv
‑
2)的合成
[0355]
在20
‑
25℃下,在装有温度计和磁力搅拌子的100ml玻璃容器中,装入:6.39g(65.76mmol,3.0eq)kscn和25ml(19.8g)干燥meoh(通过分子筛干燥)。一边搅拌,一边作为固体向混合物中加入5.06g(21.92mmol,1.0eq)d3
‑
n
‑
甲苯磺酰氧基乙脒(x
‑
2)。在20
‑
25℃下搅拌5分钟后,加入4.2ml(3.12g,24.11mmol,1.1eq)dipea。反应混合物在20
‑
25℃下搅拌过夜,逐渐变成白色悬浮液。tlc监测:ch2cl2:meoh=95:5,用kmno4显示。18小时后未完全反应,之后加热到40℃,搅拌2小时。之后,将沉淀(白色固体)过滤,用2
×
20ml meoh洗涤。向滤液中加入30ml水,之后在40℃真空(100
‑
150毫巴)下蒸发掉meoh。水性残留物用7
×
50ml etoac萃取,用na2so4将合并的有机相干燥,之后蒸发。粗产物为4.2g橙色油状物。用10ml ch2cl2处理后,将700mg粘性沉淀过滤,用5ml iproac对其进行处理,从中分离出173mg淡黄色固体(粗产率:7%)。hplc测定:63w/w%(条件b)。hplc
‑
ms(条件b)同位素纯度:73%。将ch2cl2和iproac处理的母液合并,在40℃真空下蒸发,获得2.55g粘性橙色固体。hplc测定:10w/w%(条件b)。整个反应的校正产率:14.5%(测定0.109g 0.255g)。
[0356]
实施例2:氘代3
‑
(甲基
‑
d3)
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼(i
‑
2)的合成
[0357][0358]
在所有步骤中,同位素纯度均得以保留。
[0359]
步骤1:sandmeyer溴化形成(iii
‑2‑
a)
[0360]
在20
‑
25℃下,在装有通到充满10w/w%naoh溶液的气体捕集器的出气口、滴液漏斗、温度计和磁力搅拌子的25ml玻璃容器中,装入2.7ml(4.61g溶液,含有2.85g hbr,35.2mmol,3.0eq)62w/w%hbr溶液(d:1.702g/ml)、2.1ml水和1.39g(11.76mmol,1eq)化合物(iv
‑
2)(同位素纯度:93.4%,滴定测定:85%)。将混合物加热到40℃。之后,通过滴液漏斗加入1.22g(17.64mmol,1.5eq)溶解在2ml水中的nano2,加入速率使得温度保持在40
‑
45℃下(在5分钟内)。在加入过程中,吸收器内可以观察到棕色气体的强烈鼓泡,烧瓶底部分离出油相。加入后,将混合物搅拌1小时,用tlc(hex:etoac=1:1)和hplc进行监测。反应混合物冷却到20
‑
25℃。两相体系用2
×
10ml二氯甲烷进行萃取。合并的有机相(深褐色)用5ml 5w/w%naoh溶液洗涤。将相分离。在40℃真空(300
‑
100毫巴)下将有机相蒸发,获得1.51g黄色油状物(iii
‑2‑
a)。产率:78%(1.51g)。gc(条件a):94.4%。gc
‑
ms同位素纯度:93.1%。同位素纯度没有变化。
[0361]
步骤2:乙氧羰基化形成(ii
‑2‑
b)
[0362]
在20
‑
25℃下,在惰性气氛下,向50ml高压釜中装入1.2g(6.60mmol,1.0eq)粗化合物(iii
‑2‑
a)(同位素纯度:93.1%,gc:94.9%)、12ml(10体积)无水乙醇和706mg(8.58mmol,1.3eq)naoac。之后,将氮气鼓泡通过混合物,之后加入19mg(0.033mmol,0.005mol eq)xantphos和7.5mg(0.033mmol,0.005mol eq)pd(oac)2。将烧瓶封闭,用氮气吹扫四次,再用co吹扫四次,之后装入co至4巴。在强烈搅拌下将混合物加热到65℃。反应保持在65℃,压力保持在4巴,直到反应完成(~45小时)。用gc对反应进行监测。去除co并用氮气吹扫后,在40℃真空下除去溶剂,之后加入15ml甲基环己烷,将沉淀过滤并用5ml甲基环己烷洗涤。溶剂在真空40℃(10毫巴)的旋转蒸发器上除去。经此处理后,产物(ii
‑2‑
b)以绿色油状物的形态被分离。其在下一步中使用而无需进一步纯化。产率:52%(706mg)。gc(条件a):81%。lcms(条件c)同位素纯度:93.4%。1h
‑
nmr(dmso
‑
d6):δ4.43(q,j=7.1hz,2h),1.34(t,j=7.1hz,3h)。
13
c
‑
nmr(dmso
‑
d6):179.0,175.9(d),158.4,63.3,18.2(h),14.3。同位素纯度没有下降。
[0363]
步骤3:肼解形成(i
‑
2)
[0364]
在20
‑
25℃下,在装有滴液漏斗、温度计和磁力搅拌子的10ml玻璃容器中,加入4ml ipa和177μl(181mg含100mg,3.11mmol,1.14eq)水合肼(55w/w%的水溶液,d:1.027g/ml)。
反应混合物冷却到0℃。通过滴液漏斗将595mg(2.73mmol,1.0eq)化合物(ii
‑2‑
b)(同位素纯度:93.4%,gc:81%)在2ml ipa中的溶液加入到混合物中,加入速率使得温度保持在0
‑
5℃下。加入后,在0
‑
5℃下将混合物搅拌1小时,之后在室温下过夜。用hplc对反应进行监测。完成后,将悬浮液冷却到0
‑
5℃,之后过滤,用2
×
1ml冷ipa洗涤滤饼,在35℃真空下干燥。经此处理后,产物(i
‑
2)以淡黄色固体的形态被分离。产率:72%(321mg)。hplc
‑
ms(条件d):96.4%。hplc
‑
ms(条件c)同位素纯度:93.4%。1h
‑
nmr(dmso
‑
d6):δ10.50(br,1h),4.79(br,2h)。
13
c
‑
nmr(dmso
‑
d6):δ183.4,175.0(d),157.8(d),18.0(h)。同位素纯度没有下降。
[0365]
实施例3:3
‑
甲基
‑
1,2,4
‑
噻二唑
‑5‑
碳酰肼(i
‑
1)的合成
[0366][0367]
步骤1:sandmeyer溴化形成(iii
‑1‑
a)
[0368]
使用100kg 5
‑
氨基
‑3‑
甲基
‑
1,2,4
‑
噻二唑(amtd),成功进行了sandmeyer溴化反应,产率为82%。
[0369]
将439.5kg(295l)48%hbr溶液和100.0kg amtd加入到反应器中的54kg(l)水中。将溶液加热至最大40℃,并且搅拌至所有起始材料溶解
‑
通过取样检查。如果原料没有完全溶解,那么在40℃下搅拌2小时就足够了。在另一个装置中将89.7kg亚硝酸钠溶于150kg(l)水,之后将溶液装入容器。将亚硝酸钠溶液分批装入反应器,该过程大约需要6小时,保持温度在40
‑
45℃下。加入结束一小时后取样。终点标准:起始原料<1.0%。如果原料>1.0%,则需要在反应混合物中加入额外的亚硝酸钠溶液(10
‑
20%)。如果反应混合物达到终点标准,则将其冷却至20
‑
25℃,加入399.0kg(300l)dcm。搅拌10分钟,静置10分钟后,将下层有机相装入容器。在反应器中用133.0kg(100l)dcm萃取水相。加入5%氢氧化钠溶液,将合并的有机相的ph调至10
‑
11。如果ph值超过11,则可能会形成大量水分。将混合物搅拌20分钟,静置30分钟后,将有机相分离到干燥装置中,并且在最大30℃的真空(约
‑
0.9巴)下浓缩。取样,终点标准为浓度:dcm含量最大5.0%。蒸馏结束后,需要在真空中搅拌4
‑
6小时,以达到最大5.0%的dcm含量。2.0%的dcm含量在下一步中是适宜的。若材料符合终点标准,则将其填充到pe衬里桶中。获得126.8kg化合物(iii
‑1‑
a),产率:81.6%,gc纯度(条件c):97.2%。
[0370]
步骤2:乙氧羰基化形成(ii
‑1‑
b)
[0371]
使用87.5kg化合物(iii
‑1‑
a),成功进行了乙氧羰基化反应,产率为87%。
[0372]
将87.5kg化合物(iii
‑1‑
a)加入到pe衬里桶中的44kg无水乙醇中,搅拌,直到获得澄清溶液。将383kg(484l)无水乙醇装入干燥、耐压的装置中。用n2惰性化后,加入由0.672kg乙酸钯(ii)和1.68kg xantphos在14.5kg冰醋酸中配制得到的催化剂溶液。在混合
物中加入52.5kg无水乙酸钠。再次用n2对该体系进行吹扫,并且将反应混合物加热到63
‑
67℃。向装置中加入co至4
‑
4.5bar,之后在2
‑
3小时内加入(iii
‑1‑
a)溶液。加入后,使用16公斤(20升)乙醇进行漂洗。每12小时取样一次,终点标准:起始物质(iii
‑1‑
a)最大1.0%。当反应达到终点标准时,通过catox催化剂释放压力并用n2吹扫装置6次。混合物通过生产过滤器过滤到干燥容器中
‑
用80kg(100l)无水乙醇洗涤。滤液在最大50℃下在减压下浓缩。浓缩物用120kg甲基环己烷稀释,搅拌30
‑
40分钟。溶液用生产过滤器过滤,用40kg甲基环己烷洗涤。溶剂在最大50℃下在减压下除去,产物通过分级真空蒸馏分离。获得73.5kg化合物(ii
‑1‑
b),产率:87.0%,gc纯度(条件a):96.9%。
[0373]
步骤3:肼解形成(i
‑
1)
[0374]
使用50kg化合物(ii
‑1‑
b),成功进行了肼解反应,产率为91%。
[0375]
将50.0kg化合物(ii
‑1‑
b)溶解在212.2kg(270l)异丙醇中。将26.7kg水合肼(55%水溶液)加入到340千克(432l)异丙醇中,并且将混合物冷却到0℃。将(ii
‑1‑
b)的溶液加入到冷的水合肼溶液中,该过程需要1
‑
2小时,温度保持在0
‑
5℃下。加入完成后,在0
‑
5℃下将混合物再搅拌1小时。搅拌1小时后,检查(ii
‑1‑
b)含量,若(ii
‑1‑
b)含量>1.0%,则将混合物加热到20
‑
25℃并取样监测反应进程。预期反应时间为20小时,直到达到终点标准:(ii
‑1‑
b)<1.0%。若反应达到终点标准,则将悬浮液冷却到0
‑
5℃并过滤。过滤后的材料用31.8kg(40l)0
‑
5℃异丙醇洗涤。获得37.5kg化合物(i
‑
1),产率:91%,hplc纯度(条件a):99.9%。
[0376]
实施例4:使用叔丁基亚硝酸酯进行sandmeyer溴化获得(iii
‑1‑
a)
[0377][0378]
在n2下,在schlenk瓶中装入cubr2(14g,62.68mmol,1.03eq)、无水mecn(110ml)和叔丁基亚硝酸酯(11ml,92.59mmol,1.5eq),之后将获得的深绿色混合物冷却至0
‑
5℃(冰浴)。向该混合物中加入amtd(7g,60.78mmol,1.0eq),持续1分钟。所得混合物在0
‑
5℃下搅拌30分钟,之后使其升温至室温并搅拌2小时。用25%氨水(50ml)和10%na2co3(50ml)的1:1混合物将混合物稀释,加入tbme(100ml)。将相分离,用tbme(2
×
50ml)萃取水层。合并的有机相用盐水(3
×
50ml)洗涤,用mgso4干燥,过滤,在减压(1毫巴,30℃)下浓缩至干燥,以悬浮在橙色油状物中的黄色固体的形态获得化合物(iii
‑1‑
a)(7.16g,39.99mmol,产率66%,hplc
‑
ms纯度>98%(条件d))。1h
‑
nmr(cdcl3):δ2.69(s,3h)。
[0379]
实施例5:使用叔丁基亚硝酸酯进行sandmeyer碘化获得(iii
‑1‑
b)
[0380][0381]
在装有进气口、滴液漏斗和温度计的1000ml三颈玻璃烧瓶中,装入55.0g(217.1mmol,1.0eq)碘、250ml干燥ch3cn和102.7ml(89.54g,868.4mmol,4.0eq)tbuono。将
反应混合物搅拌10分钟。在室温下,以固体的形式分批加入25.0g(217.1mmol,1.0eq)amtd。几分钟后,反应混合物(棕色)迅速加热至回流,观察到气体强烈排出,将反应冷却回室温。根据tlc,这个时间(约40分钟)足以完成反应。加入450ml 20w/w%na2so3水溶液将过量的碘淬灭,混合物变成黄色,但是静置后又变成棕色,进一步加入na2so3(50ml)水溶液使混合物变黄,但是其又变成棕色。将相分离,用tbme(4
×
150ml)萃取水相。合并的有机相用200ml盐水洗涤,用na2so4干燥,过滤并在30℃真空下蒸发。产物(iii
‑1‑
b)为棕色固体,粗产率为72.2%(校正产率为65.7%)(35.4g,hplc纯度(条件a):92.3面积%,qnmr:91w/w%)。用回流的2
‑
丙醇(2ml/粗g)通过重结晶纯化产物,产率为63%(23.9g,hplc纯度(条件a):99.3面积%,qnmr:99w/w%)。1h
‑
nmr(cdcl3):δ2.73(s,3h)。
13
c
‑
nmr(cdcl3):δ175.9,124.8,18.9。
[0382]
使用实施例3
‑
步骤2的条件(co,pd(oac)2,xantphos)或实施例9的条件(co,pd(pph3)2cl2)将碘化中间体(iii
‑1‑
b)成功转化为化合物(ii
‑1‑
a)。
[0383]
实施例6:使用亚硝酸钠进行sandmeyer碘化获得(iii
‑1‑
b)
[0384][0385]
在0℃下,将亚硝酸钠(70mg,1.01mmol)和碘化钾(215mg,1.3mmol)的水溶液(0.5ml)加入到amtd(58mg,0.50mmol)和对甲苯磺酸一水合物(305mg,1.74mmol)混合物的无水mecn(2ml)溶液中,持续5分钟(无色溶液在第一滴后变成黄色,之后变成黑色浆状,但加入结束后变成黑色溶液)。反应混合物在室温下搅拌过夜。之后用nahco3饱和溶液(15ml)稀释反应混合物,并且用etoac(3
×
10ml)萃取。合并的有机层用mgso4进行干燥,过滤并在减压(1
‑
2毫巴,30℃)下浓缩至干燥,获得149mg的深色油状物。hplc
‑
ms(条件d):90面积%。1h
‑
nmr(cdcl3):δ2.73(s,3h)。
13
c
‑
nmr(cdcl3):δ175.9,124.8,18.9。
[0386]
实施例7:通过锂交换进行乙氧羰基化
[0387]
噻二唑上烷氧羰基的引入也可以通过锂交换来进行。
[0388][0389]
在装有进气口、滴液漏斗和温度计的三颈玻璃容器中,在氩气下,10.0g(55.87mmol,1eq)化合物(iii
‑1‑
a)溶解在100ml干燥methf中。将获得的混合物冷却到
‑
65℃。之后在20分钟内滴加15.6g(22.0ml,55.87mmol)己基锂的己烷溶液(33%),温度保持在
‑
65℃下。完全加入后,取样获得的棕色反应混合物:反应混合物 氯甲酸乙酯 水 tbme。用hplc(条件b)分析tbme相。通过工艺中的控制,发现有5.5%未反应的起始原料,因此在
‑
65℃下向反应混合物中加入2.2ml(5.58mmol)己基锂的己烷溶液。20分钟后,检测到起始溴化物已完全消耗,因此通过冷却的套管将反应混合物转移到
‑
65℃下预冷却的32ml(335.16mmol,6eq)氯甲酸乙酯和30ml干燥methf的混合物中,持续15分钟。之后使获得的暗
橙色反应混合物升温至室温,之后加入200ml去离子水。将相分离,有机相用na2so4干燥并浓缩获得13.2g棕色粗产物。通过1毫巴下的真空蒸馏对八克粗产物进行纯化,收集到四个馏分。蒸馏后,将收集到的馏分合并,以无色油状物的形态获得1.9g产物(产率:29%,hplc纯度(条件b):92%)。1h
‑
nmr(cdcl3):δ4.51(q,j=7.1hz,2h),2.77(s,3h),1.44(t,j=7.1hz,3h)。
13
c
‑
nmr(cdcl3):δ178.8,175.2,158.6,63.4,19.1,14.3。
[0390]
实施例8:fe(co)5的乙氧羰基化
[0391]
烷氧羰基化在fe(co)5的存在下也成功进行。
[0392][0393]
在20
‑
25℃下,在惰性气氛下,向270ml高压釜中装入5.0g(28.0mmol,1.0eq)粗化合物(iii
‑1‑
a)(纯度:96w/w%,含3%二氯甲烷)、50.0ml(10体积)无水乙醇(水256ppm)和3.0g(36.55mmol,1.3eq)naoac(水256ppm)。使氮气鼓泡通过该混合物,持续5分钟,之后加入80.0mg(0.14mmol,0.005mol eq)xantphos、31.5mg(0.14mmol,0.005mol eq)pd(oac)2和376μl(560mg,2.86mmol,0.1eq)fe(co)5(d:1.45g/ml)。将烧瓶封闭,用氮气吹扫四次,再用co吹扫四次,之后装入co至4巴。在强烈搅拌下将混合物加热到65℃。加热过程中压力达到5巴。反应保持在65℃,压力保持在4巴,直到反应完成(~24小时)。用gc(条件a)监测反应,取样:用3.6ml乙醇稀释的0.4ml反应混合物。gc转化率:17.5小时后为59%,24小时后为84.5%。
[0394]
实施例9:用pd(pph3)2cl2催化剂进行乙氧羰基化
[0395]
在pd(pph3)2cl2催化剂的存在下,烷氧羰基化也成功进行。
[0396]
溴化中间体(iii
‑1‑
a)上的烷氧羰基化反应
[0397][0398]
在20
‑
25℃下,在惰性气氛下,在装有磁力搅拌子的800ml高压釜中,装入5.0g(27.9mmol,1.0eq)蒸馏的(iii
‑1‑
a)、140ml(28体积)无水乙醇和5ml(3.64g,35.9mmol,1.28eq)tea。该混合物用氮气吹扫10分钟,之后加入1.0g(1.42mmol,0.05eq)pd(pph3)2cl2。将烧瓶封闭,用氮气吹扫三次,再用co吹扫三次,之后装入co至5巴。在强烈搅拌下将混合物加热到100℃,压力增加到6.5巴,将其搅拌至完全反应(~31小时)。反应在8小时、14小时和31小时后用lcms(条件b)进行监测(取样:150μl反应混合物,用硅藻土过滤,用乙醇(2
×
150μl)洗涤,直接使用)。之后,反应混合物用5g硅藻土过滤,并且用2
×
50ml乙醇洗涤。在40℃真空下从滤液中蒸发掉etoh,获得12.09g棕色固体,悬浮在150ml tbme中。之后用3
×
50ml水洗涤有机相,之后在40℃真空下进行蒸发,获得油状物与固体的黄色混合物。粗产率:
82.5%(3.96g),lcms(条件b):42面积%。1h
‑
nmr:估计为66n/n%(不考虑pph3o)。
[0399]
碘化中间体(iii
‑1‑
b)上的烷氧羰基化反应
[0400][0401]
在高压釜中装入化合物(iii
‑1‑
b)(1g,4.34mmol)、tea(0.75ml,5.34mmol)、二氯双(三苯基膦)钯(ii)(60mg,0.08mmol)的无水乙醇溶液(20ml)。之后用一氧化碳吹扫容器两次,加压到10巴,加热到110℃。hplc
‑
ms监测(条件d)显示在4小时内已完全转化。混合物用硅藻土过滤,固体用etoh(20ml)洗涤,滤液在减压(1
‑
2毫巴,40℃)下浓缩至干燥,获得黄橙色固体。将后者溶于二氯甲烷(5ml)中并加入tbme(15ml),直到形成沉淀。将该灰白色固体过滤,用tbme(10ml)洗涤,滤液在高真空(2毫巴,40℃)下浓缩至干燥,获得854mg橙色固体。基于1h
‑
nmr估计纯度为59wt%(检测到pph3o和tea
‑
hi盐),校正产率为67%。
[0402]
实施例10:用pd(oac)2‑
pph3催化剂进行乙氧羰基化
[0403]
在pd(oac)2‑
pph3催化剂的存在下,烷氧羰基化也成功进行。
[0404][0405]
在20
‑
25℃下,在惰性气氛下,在装有机械搅拌的250ml高压釜中,装入3.75g(20.9mmol,1.0eq)蒸馏的(iii
‑1‑
a)、105ml(28体积)无水乙醇和3.75ml(2.73g,26.99mmol,1.29eq)tea。该混合物用氮气吹扫10分钟,之后加入578mg(1.05mmol,0.105eq)pph3和235mg(11.17mmol,0.05eq)pd(oac)2。将烧瓶封闭,用氮气吹扫三次,再用co吹扫三次,之后装入co至5巴。在强烈搅拌下将混合物加热到100℃,压力增加到6.5巴,将其搅拌至完全反应(~17小时)。反应在17小时后用hplc(条件b)进行监测(取样:150μl反应混合物,用硅藻土过滤,用乙醇(2
×
150μl)洗涤,直接使用)。之后,反应混合物用5g硅藻土过滤,并且用2
×
50ml乙醇洗涤。在40℃真空下从滤液中蒸发掉etoh,获得7.03g棕色固体,悬浮在150mltbme中。之后用3
×
50ml水洗涤有机相,之后在40℃真空下进行蒸发,获得油状物与固体的黄色混合物。粗产率:84.7%(3.05g),lcms(条件b):71面积%。1h
‑
nmr:估计为80n/n%(不考虑pph3o)。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。