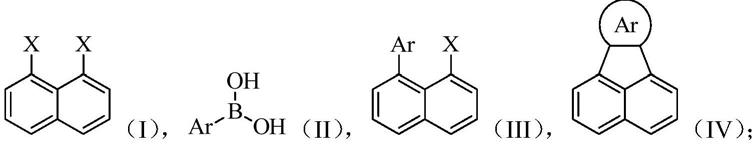
1.本发明涉及合成化学领域,具体而言,本发明涉及基于钯催化的分子内关环反应构造荧蒽及其衍生物的新方法。
背景技术:
2.荧蒽及其衍生物(取代荧蒽)是一类分子体系简单,结构明确的有机小分子,被广泛的应用于包括oled发光层、染料敏化太阳能电池构筑、荧光化学传感器、活细胞成像、电致化学发光等新兴研究领域。荧蒽的发现可以追溯到19世纪,20世纪时化学家们确定了荧蒽的分子结构,目前工业上生产无取代荧蒽的主要手段仍然是从煤焦油的高沸点组分中提取而后纯化。荧蒽衍生物的合成方法学研究可以上溯到二十世纪五六十年代,c.f.h.allen和j.a.vanallan采用knoevenagel反应首先将苊醌与β取代的羰基化合物缩合得到(取代)苊并环戊二烯酮的结构单元,而后通过diels
‑
alder反应及后续的retro
‑
diels
‑
alder或氧化脱氢反应得到荧蒽衍生物。1992年时,joseph e.rice和zhen
‑
wei cai发展了一种基于1
‑
苯基萘的三氟甲磺酸酯衍生物的分子内偶联反应,在140℃及双三苯基膦二氯化钯的催化下能够得到一系列的荧蒽衍生物。2003年时,armin de meijere等人报道了一种基于串联suzuki
‑
heck反应构建荧蒽衍生物的新方法,其基本思想是在1
‑
苯基萘的苯环1位或3位引入溴原子,而后通过heck型的反应实现分子内关环进而实现荧蒽分子骨架的构筑。peter langer等人于2011年又发展了一种合成8,9
‑
双取代荧蒽结构的新方法,他们基于heck/电环化/脱氢的串联反应策略获得了令人满意的产率及底物适用性。2014年michael j.krische课题组又报道了钌催化的苊烯二醇与二烯的[4 2]环加成而后一步脱水构造荧蒽衍生物的新策略,进一步丰富了合成荧蒽类结构的工具库。
[0003]
总体来说,以上方法学可主要分为以下几类:
[0004]
(1)苊醌缩合
‑
环加成型,这一方法学的优点在于原料廉价易得,产率相对较高,通过缩合反应能够实现荧蒽7号位和10号位的官能团化,通过diels
‑
alder反应底物的选择,能够实现8号位和9号位的官能团化,而通过对底物苊醌的前修饰,又能够实现3号位和4号位的取代。其缺陷主要体现在对于8号位,9号位短烷基链取代或卤素取代的荧蒽结构的合成无能为力;对于7号位,10号位卤素取代的荧蒽衍生物合成同样无法实现;基于此策略合成芳基[k]并荧蒽结构存在困难。此外不同β取代基羰基化合物的合成并不是一项容易的工作,在后续diels
‑
alder环加成反应中常用到的二甲苯类溶剂毒性较大,具有一定的风险。
[0005]
(2)取代1
‑
苯基萘关环偶联型,这一方法学对于芳基并荧蒽结构的合成较为有效,然而无论是三氟甲磺酸酯取代或溴代1
‑
苯基萘的合成都经历比较复杂的步骤,因此该方法学底物适用性相当有限。在最终的催化关环反应中,均需要采取较为严苛的反应条件(100℃以上高温)以及使用贵金属催化剂,也是很大的限制。
[0006]
(3)基于其他苊类衍生物的方法学,其优势在于底物适用性更加广泛,对于荧蒽衍生物的合成方法学而言是极为重要的丰富,但这几种反应体系,使用的催化剂均非常昂贵,其应用暂时被限制在实验室级别的制备,有待进一步的条件优化和催化剂开发来实现大规
模的生产。
[0007]
综上所述,现有的制备荧蒽及其衍生物的方法仍有待改进。
技术实现要素:
[0008]
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明的一个目的在于提出基于钯催化的分子内关环反应构造荧蒽及其衍生物的的新方法。该方法以萘的1号、8号位卤素取代物为底物,与芳基硼酸经两步反应合成荧蒽及其衍生物,底物获取简单、适用范围宽,且容易调控荧蒽衍生物的7号、8号、9号、10号取代位,为最终的目标分子结构提供了广泛的可能性。
[0009]
在本发明的一个方面,本发明提出了一种制备荧蒽及其衍生物的方法,其特征在于,包括:
[0010]
(1)使式i所示化合物与式ii所示化合物发生偶联反应,得到式iii所示化合物;
[0011]
(2)使式iii所示化合物发生分子内关环反应,得到式iv所示化合物;
[0012][0013]
其中,x为卤素原子,ar为取代或未取代的芳基。
[0014]
根据本发明上述实施例的制备荧蒽及其衍生物的方法,首先利用萘的1号、8号位卤素取代物(式i所示化合物)与芳基硼酸(式ii所示化合物)偶联得到萘的芳基取代物(式iii所示化合物)。其中,式ii所示化合物种的ar为取代或未取代的芳基,通过调控该芳基中的取代基,即可调控制备得到的荧蒽及其衍生物中7号、8号、9号、10号位的取代基(可以理解的是,当ar为未取代的苯基时,制备得到的产物为荧蒽;当ar为取代的苯基、取代或未取代的其他芳基时,制备得到的产物为荧蒽衍生物),为最终的目标分子结构提供了广泛的可能性。本发明提供的方法中,反应底物廉价易得、适用范围宽,且涉及的反应条件温和,容易大规模制备。
[0015]
另外,根据本发明上述实施例的制备荧蒽及其衍生物的方法还可以具有如下附加的技术特征:
[0016]
在本发明的一些实施例中,x为cl或br,ar为取代或未取代的c4~c16芳基。
[0017]
在本发明的一些实施例中,所述c4~c16芳基为苯基、萘基、蒽基、菲基、呋喃基、吡喃基、吡咯基、吡啶基或噻吩基。
[0018]
在本发明的一些实施例中,所述c4~c16芳基任选地被c1~c4烷基、c1~c4烷氧基、卤素原子中的至少之一取代。
[0019]
在本发明的一些实施例中,所述偶联反应在第一溶剂中进行,所述第一溶剂选自甲苯、水、四氢呋喃、乙醇、苯、乙腈、1,4
‑
二氧六环中的至少之一。
[0020]
在本发明的一些实施例中,所述偶联反应中,式i所示化合物与式ii所示化合物的摩尔比为1:(1.05~1.2)。
[0021]
在本发明的一些实施例中,所述偶联反应在第一钯催化剂存在的条件下进行,所述第一钯催化剂选自pd(pph3)4、pd(dppf)cl2、pd(oac)2、pd(pph3)2cl2中的至少之一。
[0022]
在本发明的一些实施例中,所述偶联反应在第一添加剂存在的条件下进行,所述第一添加剂选自四丁基溴化铵、碳酸钾、碳酸钠、磷酸钾、氟化铯、叔丁醇钠中的至少之一。
[0023]
在本发明的一些实施例中,所述四丁基溴化铵的添加量为8mol%~12mol%;
[0024]
在本发明的一些实施例中,所述碳酸钾的添加量为225mol%~275mol%。
[0025]
在本发明的一些实施例中,所述偶联反应在80℃~100℃下进行45h~50h。
[0026]
在本发明的一些实施例中,所述分子内关环反应在第二溶剂中进行,所述第二溶剂选自二甲基亚砜、1,4
‑
二氧六环、乙腈、四氢呋喃、异丙醇中的至少之一。
[0027]
在本发明的一些实施例中,所述分子内关环反应在第二钯催化剂存在的条件下进行,所述第二钯催化剂选自pd(dppf)cl2、pd(pph3)4、pd(oac)2、pd(pph3)2cl2中的至少之一。
[0028]
在本发明的一些实施例中,所述分子内关环反应在第二添加剂存在的条件下进行,所述第二添加剂选自双(频哪醇合)二硼、醋酸钾、碳酸钾、碳酸钠、磷酸钾、氟化铯、叔丁醇钠中的至少之一。
[0029]
在本发明的一些实施例中,所述双(频哪醇合)二硼的添加量为100mol%~120mol%;
[0030]
在本发明的一些实施例中,所述醋酸钾的添加量为280mol%~320mol%。
[0031]
在本发明的一些实施例中,所述分子内关环反应在80℃~100℃下进行10h~14h。
[0032]
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
具体实施方式
[0033]
下面详细描述本发明的实施例。下面描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0034]
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
[0035]
另外,需要说明的是,本发明中对于某物质添加量摩尔百分比(mol%)的描述,是指该物质相对于式i所示化合物的摩尔百分比。
[0036]
在本发明的一个方面,本发明提出了一种制备荧蒽及其衍生物的方法,其特征在于,包括:
[0037]
(1)使式i所示化合物与式ii所示化合物发生偶联反应,得到式iii所示化合物;
[0038]
(2)使式iii所示化合物发生分子内关环反应,得到式iv所示化合物;
[0039][0040]
其中,x为卤素原子,ar为取代或未取代的芳基。
[0041]
根据本发明的一些实施例,x可以为cl或br,优选地,x为br,即式i所示化合物为1,8
‑
二溴萘。
[0042]
根据本发明的一些实施例,ar可以为取代或未取代的c4~c16芳基。通过调控ar中取代基的选择,即可调控制备得到的荧蒽及其衍生物中7号、8号、9号、10号位的取代基,从而为最终的目标分子结构提供了广泛的可能性。
[0043]
根据本发明的一些实施例,c4~c16芳基可以为苯基、萘基、蒽基、菲基、呋喃基、吡喃基、吡咯基、吡啶基或噻吩基。
[0044]
根据本发明的一些实施例,上述c4~c16芳基可以任选地被c1~c4烷基、c1~c4烷氧基、卤素原子中的至少之一取代。c1~c4烷基的具体示例包括甲基、乙基、丙基、丁基,c1~c4烷氧基的具体示例包括甲氧基、乙氧基、丙氧基、丁氧基,卤素原子的具体示例包括f、cl、br、i。
[0045]
根据本发明的一些实施例,偶联反应在第一溶剂中进行,第一溶剂可以选自甲苯、水、四氢呋喃、乙醇、苯、乙腈、1,4
‑
二氧六环中的至少之一,优选为甲苯和水的混合溶剂。
[0046]
根据本发明的一些实施例,偶联反应中,式i所示化合物与式ii所示化合物的摩尔比为1:(1.05~1.2),例如可以为1:1.05、1:1.06、1:1.08、1:1.1、1:1.15、1:1.2等,优选为1:1.1。
[0047]
根据本发明的一些实施例,偶联反应在第一钯催化剂存在的条件下进行,第一钯催化剂可以选自pd(pph3)4、pd(dppf)cl2、pd(oac)2、pd(pph3)2cl2中的至少之一,优选为pd(pph3)4。
[0048]
根据本发明的一些实施例,偶联反应在第一添加剂存在的条件下进行,第一添加剂可以选自四丁基溴化铵、碳酸钾、碳酸钠、磷酸钾、氟化铯、叔丁醇钠中的至少之一,优选为四丁基溴化铵和碳酸钾。
[0049]
根据本发明的一些实施例,四丁基溴化铵的添加量可以为8mol%~12mol%,例如8mol%、9mol%、10mol%、11mol%、12mol%等,优选为10mol%;如果四丁基溴化铵的添加量过低,则可能反应产率大大降低;如果四丁基溴化铵的添加量过高,则可能在体系中产生大量不溶物,增加后处理难度,并且相应提高反应成本。
[0050]
根据本发明的一些实施例,碳酸钾的添加量可以为225mol%~275mol%,例如225mol%、235mol%、245mol%、250mol%、255mol%、265mol%、275mol%等,优选为250mol%。如果碳酸钾的添加量过低,则可能反应产率大大降低;如果碳酸钾的添加量过高,则可能体系中产生大量不溶物,增加后处理难度,并且相应提高反应成本。
[0051]
根据本发明的一些实施例,偶联反应在80℃~100℃下进行45h~50h。具体的,反应温度可以为80℃、85℃、90℃、95℃、100℃等,反应时间可以为45h、46h、47h、48h、49h、50h等,优选在90℃下进行48h。如果反应温度过高,则可能引发反应物热分解,造成不可控的安全事故。
[0052]
根据本发明的一些实施例,偶联反应在惰性气体氛围中进行,例如可以在氮气氛围中进行。
[0053]
根据本发明的一些实施例,分子内关环反应在第二溶剂中进行,第二溶剂可以选自二甲基亚砜、1,4
‑
二氧六环、乙腈、四氢呋喃、异丙醇中的至少之一,优选为二甲基亚砜。
[0054]
根据本发明的一些实施例,分子内关环反应在第二钯催化剂存在的条件下进行,
第二钯催化剂可以选自pd(dppf)cl2、pd(pph3)4、pd(oac)2、pd(pph3)2cl2中的至少之一中的至少之一,优选为pd(dppf)cl2。
[0055]
根据本发明的一些实施例,分子内关环反应在第二添加剂存在的条件下进行,第二添加剂可以选自双(频哪醇合)二硼、醋酸钾、碳酸钾、碳酸钠、磷酸钾、氟化铯、叔丁醇钠中的至少之一,优选为双(频哪醇合)二硼和醋酸钾。
[0056]
根据本发明的一些实施例,双(频哪醇合)二硼的添加量可以为100mol%~120mol%,例如100mol%、105mol%、110mol%、115mol%、120mol%等,优选为110mol%。如果双(频哪醇合)二硼的添加量过低,则可能反应产率大大降低;如果双(频哪醇合)二硼的添加量过高,则可能增加后处理难度及反应成本。
[0057]
根据本发明的一些实施例,醋酸钾的添加量可以为280mol%~320mol%,例如280mol%、290mol%、300mol%、310mol%、320mol%等,优选为300mol%。如果醋酸钾的添加量过低,则可能反应产率大大降低;如果醋酸钾的添加量过高,则可能在体系中产生大量不溶物,增加后处理难度,并且相应提高反应成本。
[0058]
根据本发明的一些实施例,分子内关环反应在80℃~100℃下进行10h~14h。具体的,反应温度可以为80℃、85℃、90℃、95℃、100℃等,反应时间可以为10h、11h、12h、13h、14h等,优选在90℃下进行12h。如果反应温度过高,则可能引发反应物热分解,造成不可控的安全事故。
[0059]
根据本发明的一些实施例,分子内关环反应在惰性气体氛围中进行,例如可以在氮气氛围中进行。
[0060]
综上所述,根据本发明实施例的制备荧蒽及其衍生物的方法至少具有以下优点:
[0061]
(1)底物获取便捷:我们所发展的新方法采取的主要底物为1,8
‑
二溴萘及(取代)芳基硼酸,其中1,8
‑
二溴萘已经商品化,廉价易得;(取代)芳基硼酸有大量商品化的选择,并且合成路径成熟。相较现有技术而言,如果采取三氟甲磺酸酯取代的1
‑
苯基萘进行关环偶联获取荧蒽衍生物,首先三氟甲磺酸酯取代的1
‑
苯基萘需要多步合成才能够获取(尚未实现商品化),其次适用的底物范围很窄,能够构筑的荧蒽衍生物仅限于单取代。而如采取溴取代的1
‑
苯基萘进行关环偶联获取荧蒽衍生物,其关键底物邻二溴代芳烃或邻溴代芳基硼酸均是合成上具有困难的分子,这也就天然的限制了这种方法的底物范围。
[0062]
(2)底物适用范围宽:如上所述,由于多种(取代)芳基硼酸能够直接购买或通过锂试剂快速获取,并且经过验证,大部分底物都能够最终实现偶联得到取代荧蒽的结构。而相似的现有技术,由于上面提到的这些问题,很难有广泛的底物适用性。
[0063]
(3)反应条件温和:本发明的方法涉及到两步化学反应过程,其反应温度均在100℃以下,使用到的添加剂(四丁基溴化铵、碳酸钾、醋酸钾等)及溶剂(甲苯、水、二甲基亚砜等)均为低毒性。对于现有技术中三氟甲磺酸酯取代的1
‑
苯基萘关环偶联的方法,需要用到三氟甲磺酸酐这类危险化学品以及140℃的高温。对于现有技术中溴取代的1
‑
苯基萘关环偶联的方法,同样需要使用150℃左右的高温。
[0064]
(4)适合大规模制备:由于具备上述优势,基于本发明方法可实现了一系列荧蒽衍生物的克级制备,并且保持较高的产率。而现有技术均停留在毫克级产量,距离实际应用还有一定的障碍。
[0065]
下面参考具体实施例,对本发明进行描述,需要说明的是,这些实施例仅仅是描述
性的,而不以任何方式限制本发明。
[0066]
实施例1
[0067]
x=br
[0068]
步骤1:在圆底烧瓶中放置一枚磁子,依次加入1,8
‑
二溴萘(5.72g,20mmol),苯硼酸(2.68g,22mmol)、无水碳酸钾(6.91g,50mmol)、四丁基溴化铵(665mg,2.0mmol)及四三苯基膦钯(116mg,0.10mmol)。将圆底烧瓶进行氮气置换,加入80ml甲苯及25ml去离子水,置于90℃的油浴下反应48h。而后降至室温,加入一定量的去离子水,以乙酸乙酯萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物s1。使用柱色谱进一步提纯得中间产物s1。
[0069]
步骤2:在配置有磁子的圆底烧瓶中,依次加入中间产物s1(5.66g,20mmol),双(频哪醇合)二硼(5.59g,22mmol),pd(dppf)cl2(439mg,0.6mmol)、醋酸钾(5.89g,60mmol)。氮气置换后加入120ml二甲基亚砜,置于90℃的油浴下反应12h。而后降至室温,加入一定量的去离子水,以二氯甲烷萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物p1。采取柱色谱进一步提纯得目标产物p1。
[0070]
表征:
[0071]
s1:
[0072]
1h
‑
nmr(400mhz,chloroform
‑
d)δ=7.86
–
7.83(m,1h),7.82(t,j=1.6hz,1h),7.75(dd,j=7.4,1.1hz,1h),7.48
–
7.43(m,1h),7.40(dd,j=7.1,1.5hz,1h),7.36(dd,j=4.2,2.4hz,3h),7.31(dd,j=6.6,3.0hz,2h),7.27
–
7.20(m,1h).
[0073]
13c
‑
nmr(101mhz,chloroform
‑
d)δ=142.99,140.53,136.22,133.90,131.36,130.37,129.76,129.05,129.01,127.57,127.08,126.19,125.46,120.30.
[0074]
p1:
[0075]
1h
‑
nmr(400mhz,dmso
‑
d6)δ=8.07(d,j=6.9hz,2h),8.00(dd,j=5.5,3.1hz,2h),7.91(d,j=8.2hz,2h),7.66(dd,j=8.0,7.1hz,2h),7.38(dd,j=5.5,3.1hz,2h).
[0076]
13c
‑
nmr(101mhz,dmso
‑
d6)δ=139.24,136.68,131.95,130.11,128.79,128.31,127.37,122.42,121.30.
[0077]
实施例2
[0078]
x=br
[0079]
步骤1:在圆底烧瓶中放置一枚磁子,依次加入1,8
‑
二溴萘(5.72g,20mmol),3,4
‑
二甲氧基苯硼酸(4.00g,22mmol)、无水碳酸钾(6.91g,50mmol)、四丁基溴化铵(665mg,2.0mmol)及四三苯基膦钯(116mg,0.10mmol)。将圆底烧瓶进行氮气置换,加入80ml甲苯及25ml去离子水,置于90℃的油浴下反应48h。而后降至室温,加入一定量的去离子水,以乙酸乙酯萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物s2。使用柱色谱进一步提纯得中间产物s2。
[0080]
步骤2:在配置有磁子的圆底烧瓶中,依次加入中间产物s2(6.86g,20mmol),双(频哪醇合)二硼(5.59g,22mmol),pd(dppf)cl2(439mg,0.6mmol)、醋酸钾(5.89g,60mmol)。氮气置换后加入120ml二甲基亚砜,置于90℃的油浴下反应12h。而后降至室温,加入一定量的去离子水,以二氯甲烷萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物p2。采取柱色谱进一步提纯得目标产物p2。
[0081]
表征:
[0082]
s2:
[0083]
1h
‑
nmr(400mhz,dmso
‑
d6)δ=8.00(dd,j=11.5,8.2hz,2h),7.79(d,j=7.4hz,1h),7.54(t,j=7.6hz,1h),7.45
–
7.40(m,1h),7.36(t,j=7.8hz,1h),6.94(d,j=8.2hz,1h),6.81(d,j=1.8hz,1h),6.75(dd,j=8.1,1.9hz,1h),3.77(s,3h),3.68(s,3h).
[0084]
13c
‑
nmr(101mhz,dmso
‑
d6)δ=148.65,148.33,140.00,136.38,135.07,134.28,131.78,129.73,129.41,129.32,126.97,126.13,122.73,119.72,114.68,111.45,56.01,55.99.
[0085]
p2:
[0086]
1h
‑
nmr(400mhz,dmso
‑
d6)δ=7.97(d,j=6.9hz,2h),7.80(d,j=8.2hz,2h),7.68(s,2h),7.63
–
7.57(m,2h),3.87(s,6h).
[0087]
13c
‑
nmr(101mhz,dmso
‑
d6)δ=149.67,137.29,132.29,131.96,129.75,128.65,126.36,120.42,106.71,56.40.
[0088]
实施例3
[0089]
x=br
[0090]
步骤1:在圆底烧瓶中放置一枚磁子,依次加入1,8
‑
二溴萘(5.72g,20mmol),2
‑
萘硼酸(3.78g,22mmol)、无水碳酸钾(6.91g,50mmol)、四丁基溴化铵(665mg,2.0mmol)及四三苯基膦钯(116mg,0.10mmol)。将圆底烧瓶进行氮气置换,加入80ml甲苯及25ml去离子水,置
于90℃的油浴下反应48h。而后降至室温,加入一定量的去离子水,以乙酸乙酯萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物s3。使用柱色谱进一步提纯得中间产物s3。
[0091]
步骤2:在配置有磁子的圆底烧瓶中,依次加入中间产物s3(6.66g,20mmol),双(频哪醇合)二硼(5.59g,22mmol),pd(dppf)cl2(439mg,0.6mmol)、醋酸钾(5.89g,60mmol)。氮气置换后加入120ml二甲基亚砜,置于90℃的油浴下反应12h。而后降至室温,加入一定量的去离子水,以二氯甲烷萃取三次,将合并的有机相用卤水洗涤,分液后加入无水硫酸钠干燥,减压蒸馏得到粗产物p3。采取柱色谱进一步提纯得目标产物p3。
[0092]
表征:
[0093]
s3:
[0094]
1h
‑
nmr(400mhz,dmso
‑
d6)δ=8.10
–
8.03(m,2h),7.91(dt,j=11.8,7.2hz,3h),7.82
–
7.77(m,2h),7.63
–
7.57(m,1h),7.53
–
7.47(m,3h),7.44
–
7.37(m,2h).
[0095]
13c
‑
nmr(101mhz,dmso
‑
d6)δ=140.53,139.98,136.45,134.34,133.12,132.59,132.20,129.85,129.70,129.41,129.35,128.52,128.45,128.11,127.19,127.12,126.78,126.48,126.24,119.68.
[0096]
p3:
[0097]
1h
‑
nmr(400mhz,dmso
‑
d6)δ=8.52(s,2h),8.17(d,j=6.9hz,2h),8.00(dq,j=6.4,3.0hz,2h),7.94(d,j=8.2hz,2h),7.72(dd,j=8.0,7.1hz,2h),7.52(dt,j=6.2,3.3hz,2h).
[0098]
13c
‑
nmr(101mhz,dmso
‑
d6)δ=137.53,136.55,134.81,133.56,130.61,129.21,129.10,126.96,126.86,121.07,120.44.
[0099]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0100]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例
性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。