1.本发明涉及多肽合成技术领域,具体涉及一种利那洛肽的制备方法。
背景技术:
2.利那洛肽(linaclotide)是由美国ironwood公司研发的一种鸟苷酸环化酶c(gc
‑
c)激动剂,商品名为linzess。该药为胶囊剂,用于治疗便秘肠应激综合征(ibs
‑
c)和慢性特发性便秘(cic),它是首个具有此种作用机制的治疗便秘的药物。利那洛肽口服进入人体后通过与肠道中的鸟苷酸环化酶c型受体(gc
‑
c)结合,使细胞内和细胞外环鸟苷酸(cgmp)浓度升高,刺激肠液分泌,加快胃肠道移行,从而增加排便频率,由于其独一无二的作用机制,很有希望成为一种不但可以治疗便秘,还可以治疗腹胀、腹痛和其它临床症状的治疗药物。其结构序列如下:
[0003][0004]
现有的利那洛肽制备方法主要是两种方法:第一种:使用fmoc
‑
cys(trt)
‑
oh为起始物料,合成线肽后,在一定的浓度、温度、溶液体系中一步氧化得到利那洛肽;第二种:cys的保护基团分别使用trt、acm、mmt、pmeobzl形成对保护cys侧链巯基,并逐步去除cys上的保护基团定向形成二硫键。
[0005]
方法1得到线肽后一步氧化制备利那洛肽,虽然一步反应即得到了三对二硫键,但是存在弊端:产物可靠性较差,自然氧化不能确定三对二硫键的实际位置导致目标产物产率低;方法2虽然在生产过程中定向形成了二硫键,能得到比较可靠的产品,但是也存在弊端:各个保护基团逐步去除过程反应体系复杂,最终目标产品存在有害物质种类增加、产率低的问题。
技术实现要素:
[0006]
鉴于此,本发明的目的在于提供一种利那洛肽的制备方法,本发明提供的制备方法收率和纯度高。
[0007]
为了实现上述发明目的,本发明提供以下技术方案:
[0008]
本发明提供了一种利那洛肽的制备方法,包括以下步骤:
[0009]
(1)将肽树脂、带保护基团的胱氨酸化合物、偶联试剂和偶联反应溶剂混合进行第一偶联反应,得到第一中间体,所述第一中间体的结构如式i所示:
[0010][0011]
所述带保护基团的胱氨酸化合物的结构式如式vi所示:
[0012][0013]
所述式vi中,x1为dde、alloc、boc或mtt,所述r1为oall或odmab;
[0014]
所述肽树脂的结构式如式vii所示:
[0015]
h
‑
asn(trt)
‑
pro
‑
ala
‑
cys(acm)
‑
thr(tbu)
‑
gly
‑
cys(trt)
‑
tyr(tbu)
‑
resin式vii;
[0016]
(2)按照所述步骤(1),在所述第一中间体代替所述肽树脂,以剩余保护氨基酸依次替代所述带保护基团的胱氨酸化合物进行偶联反应,得到第二中间体,所述第二中间体的结构如式ii所示:
[0017][0018]
所述剩余的保护氨基酸依次为:fmoc
‑
cys(trt)
‑
oh、fmoc
‑
tyr(tbu)
‑
oh、fmoc
‑
glu(otbu)
‑
oh和fmoc
‑
cys(acm)
‑
oh;
[0019]
(3)将所述第二中间、脱保护试剂和催化剂混合脱除保护基团r1得到第三中间体,所述第三中间体的结构如式iii所示:
[0020][0021]
(4)按照所述步骤(1),所述第三中间体发生分子内偶联反应,得到第四中间体,所述第四中间体的结构如式iv所示:
[0022][0023]
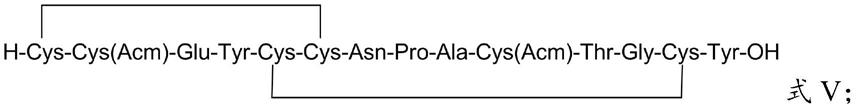
(5)将所述第四中间体在酸性裂解液中进行裂解,得到第五中间体,所述第五中间体的结构如式v所示:
[0024][0025]
(6)将所述第五中间体、酸性溶剂和碘氧化试剂混合进行氧化反应,得到所述利那洛肽。
[0026]
优选的,步骤(3)中,当所述保护基团r1为oall时,所述脱除保护试剂为醋酸、nmm和dcm的混合物,所述催化剂为钯催化剂;当所述保护基团r1为odmab时,所述脱除保护试剂为水合肼的dmf溶液。
[0027]
优选的,步骤(5)中,所述酸性裂解液为tfa和dmso的混合溶液,所述tfa和dmso的混合溶液中,所述dmso的体积浓度为5~30%。
[0028]
优选的,步骤(5)中,所述第四中间体的质量和酸性裂解液的体积比为(10~13)g:150ml;
[0029]
所述裂解的温度为20~40℃,所述裂解的时间为1.5~3h。
[0030]
优选的,所述碘氧化试剂为碘的甲醇溶液或碘的乙腈溶液,所述碘氧化试剂中碘的质量浓度为30~35g/l。
[0031]
优选的,步骤(6)中,所述第五中间体、酸性溶剂和碘氧化试剂混合得到的混合液中,所述第五中间体的质量浓度为1~10mg/ml。
[0032]
优选的,所述氧化反应的温度为5~45℃,所述氧化反应的时间为10~20min。
[0033]
优选的,步骤(1)中,所述肽树脂和带保护基团的胱氨酸化合物的物质的量之比为1:(2~4)。
[0034]
优选的,步骤(1)中,所述肽树脂采用fmoc固相合成法得到,合成所述肽树脂的起始树脂包括ctc树脂或wang树脂;
[0035]
所述起始树脂的取代度为0.65~1.2mmol/g。
[0036]
优选的,步骤(1)和步骤(2)中,所述偶联反应的温度独立的为20~40℃,所述偶联反应的时间独立地为1.2~2h。
[0037]
本发明提供了一种利那洛肽的制备方法,包括以下步骤:(1)将肽树脂、带保护基团的胱氨酸化合物、偶联试剂和偶联反应溶剂混合进行第一偶联反应,得到第一中间体;(2)按照所述步骤(1),在所述第一中间体代替所述肽树脂,以剩余保护氨基酸依次替代所述带保护基团的胱氨酸化合物进行偶联反应,得到第二中间体;所述剩余的保护氨基酸依次为:fmoc
‑
cys(trt)
‑
oh、fmoc
‑
tyr(tbu)
‑
oh、fmoc
‑
glu(otbu)
‑
oh和fmoc
‑
cys(acm)
‑
oh;(3)将所述第二中间、脱保护试剂和催化剂混合脱除保护基团r1得到第三中间体,(4)按照所述步骤(1),所述第三中间体发生分子内偶联反应,得到第四中间体;(5)将所述第四中间体在酸性裂解液中进行裂解,得到第五中间体;(6)将所述第五中间体、酸性溶剂和碘氧化试剂混合进行氧化,得到所述利那洛肽。本发明提供的制备方法,采用如式vi所示结构的带保护基团的胱氨酸化合物与肽树脂进行偶联反应,然后偶联剩余氨基酸后通过脱除保护基团直接完成一步二硫键的定向搭桥,在随后的裂解过程中完成一步二硫键的定向搭桥;裂解得到中间产物v后,仅用碘对剩余的一对acm基团保护的巯基进行定向搭桥得到利那洛肽。本发明定向形成三对二硫键,保证了最终产品结构的正确性且具有高收率和高纯度。
[0038]
本发明提供的制备方法操作流程简单、产品收率高,有利规模化生产。
附图说明
[0039]
图1为实施例1制备的利那洛肽的hplc谱图。
具体实施方式
[0040]
本发明提供了一种利那洛肽的制备方法,包括以下步骤:
[0041]
(1)将肽树脂、带保护基团的胱氨酸化合物、偶联试剂和偶联反应溶剂混合进行第一偶联反应,得到第一中间体,所述第一中间体的结构如式i所示:
[0042][0043]
所述带保护基团的胱氨酸化合物的结构式如式vi所示:
[0044][0045]
所述式vi中,x1为dde、alloc、boc或mtt,所述r1为oall或odmab;
[0046]
所述肽树脂的结构式如式vii所示:
[0047]
h
‑
asn(trt)
‑
pro
‑
ala
‑
cys(acm)
‑
thr(tbu)
‑
gly
‑
cys(trt)
‑
tyr(tbu)
‑
resin式vii;
[0048]
(2)按照所述步骤(1),在所述第一中间体代替所述肽树脂,以剩余保护氨基酸依次替代所述带保护基团的胱氨酸化合物进行偶联反应,得到第二中间体,所述第二中间体的结构如式ii所示:
[0049][0050]
所述剩余的保护氨基酸依次为:fmoc
‑
cys(trt)
‑
oh、fmoc
‑
tyr(tbu)
‑
oh、fmoc
‑
glu(otbu)
‑
oh和fmoc
‑
cys(acm)
‑
oh;
[0051]
(3)将所述第二中间体、脱保护试剂和催化剂混合脱除保护基团r1得到第三中间体,所述第三中间体的结构如式iii所示:
[0052][0053]
(4)按照所述步骤(1),所述第三中间体发生分子内偶联反应,得到第四中间体,所述第四中间体的结构如式iv所示:
[0054][0055]
(5)将所述第四中间体在酸性裂解液中进行裂解,得到第五中间体,所述第五中间体的结构如式v所示:
[0056][0057]
(6)将所述第五中间体、酸性溶剂和碘氧化试剂混合进行氧化反应,得到所述利那洛肽。
[0058]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0059]
在本发明中,缩写代表的物质如表1所示:
[0060]
表1缩写对应的物质名称
[0061]
[0062]
[0063][0064]
本发明将肽树脂、带保护基团的胱氨酸化合物、偶联试剂和偶联反应溶剂混合进行第一偶联反应,得到第一中间体,所述第一中间体的结构如式i所示:
[0065][0066]
所述带保护基团的胱氨酸化合物的结构式如式vi所示:
[0067][0068]
所述式vi中,x1为dde、alloc、boc或mtt,所述r1为oall或odmab;
[0069]
所述肽树脂的结构式如式vii所示:
[0070]
h
‑
asn(trt)
‑
pro
‑
ala
‑
cys(acm)
‑
thr(tbu)
‑
gly
‑
cys(trt)
‑
tyr(tbu)
‑
resin式vii。
[0071]
在本发明中,所述肽树脂的结构式如式vii所示:h
‑
asn(trt)
‑
pro
‑
ala
‑
cys(acm)
‑
thr(tbu)
‑
gly
‑
cys(trt)
‑
tyr(tbu)
‑
resin式vii。在本发明中,所述肽树脂优选采用fmoc固相合成法得到。
[0072]
在本发明中,所述肽树脂的制备方法优选包括以下步骤:
[0073]
1)将起始树脂、fmoc
‑
tyr(tbu)
‑
oh、缚酸剂和取代反应溶剂混合进行取代反应,得到取代树脂;
[0074]
2)将所述取代树脂、fmoc
‑
cys(trt)
‑
oh、偶联试剂和偶联反应溶剂混合进行第i偶联反应,得到第i偶联树脂;
[0075]
3)按照步骤2),在所述第i偶联树脂代替所述取代树脂,以肽树脂结构中所需的剩余保护氨基酸依次替代所述fmoc
‑
cys(trt)
‑
oh进行偶联反应,得到所述肽树脂,所述肽树脂结构中所需的剩余保护氨基酸依次为fmoc
‑
gly
‑
oh、fmoc
‑
thr(tbu)
‑
oh、fmoc
‑
cys(acm)
‑
oh、fmoc
‑
ala
‑
oh、fmoc
‑
pro
‑
oh和fmoc
‑
asn(trt)
‑
oh。
[0076]
本发明将起始树脂、fmoc
‑
tyr(tbu)
‑
oh、缚酸剂和取代反应溶剂混合进行取代反应,得到取代树脂。
[0077]
在本发明中,合成所述肽树脂的起始树脂优选包括ctc树脂或wang树脂,更优选包括ctc树脂;所述起始树脂的取代度优选为0.65~1.2mmol/g,更优选为0.66~1.1mmol/g。
[0078]
在本发明中,所述缚酸剂优选为dipea或diea,更优选为dipea。
[0079]
在本发明中,所述取代溶剂优选为n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺或二氯
甲烷,更优选为二氯甲烷。
[0080]
在本发明中,所述起始树脂和fmoc
‑
tyr(tbu)
‑
oh的物质的量之比优选为1:(1.5~5),更优选为1:(1.5~2)。
[0081]
在本发明中,所述fmoc
‑
tyr(tbu)
‑
oh的质量和缚酸剂的体积之比为(1~2.5)g:(1~2)ml,优选为(1.1~2.3)g:(1.1~1.9)ml。
[0082]
本发明对所述取代反应溶剂的用量没有特殊要求,能够时所述fmoc
‑
tyr(tbu)
‑
oh完全溶剂即可。
[0083]
在本发明中,所述起始树脂、fmoc
‑
tyr(tbu)
‑
oh、缚酸剂和取代反应溶剂的混合顺优选为:将所述起始树脂、fmoc
‑
tyr(tbu)
‑
oh和所述取代反应溶剂进行第一混合,得到第一混合液,将所述第一混合液和缚酸剂进行第二混合。
[0084]
在本发明中,所述取代反应的温度优选为20~30℃,更优选为25℃,所述取代反应的时间优选为2~5h,更优选为3h。在本发明中,所述取代反应优选在搅拌的条件下进行,本发明对所述搅拌的具体实施过程没有特殊要求。
[0085]
所述取代反应结束后,本发明优选对所述取代反应体系进行后处理,得到所述取代树脂。在本发明中,所述后处理优选包括:依次进行封端所述取代反应液中未反应的初始树脂、固液分离得到固体产品、洗涤和干燥。
[0086]
在本发明中,所述封端优选为向所述取代反应液中加入封端试剂进行封端反应,所述封端试剂优选为甲醇,所示甲醇优选为色谱级甲醇;在本发明中,所述封端试剂的体积和初始树脂的质量比优选为200ml:200g,在本发明中,所述封端试剂加入的温度优选为室温;所述封端试剂加入后封端反应的时间优选为25~40min,更优选为30min。在本发明中,所述过固液分离的方式优选为抽滤,本发明对所述抽滤的具体实施方案没有特殊要求。本发明优选对所述固液分离得到的固体产品进行洗涤,得到所述取代树脂;在本发明中,所述洗涤优选包括依次进行:dmf洗涤、甲醇洗涤、dcm洗涤和甲醇洗涤,在本发明中,所述dmf洗涤的次数优选为2次,本发明对所述dmf的用量没有特殊要求,所述甲醇洗涤的次数优选为2次,本发明对所述甲醇的用量没有特殊要求;所述dcm洗涤的次数优选为2次,本发明对所述dcm的用量没有特殊要求,所述甲醇洗涤的次数优选为2次,本发明对所述甲醇的用量没有特殊要求。本发明优选固液分离去除甲醇洗涤剂后对所述固体产品进行干燥,在本发明中,所述干燥优选为真空干燥,所述真空干燥的温度优选为40℃,干燥至固体产品失重<3%停止干燥,得到所述取代树脂。
[0087]
在本发明中,所述取代树脂的取代度优选为0.4~0.8mmol/g。
[0088]
得到取代树脂后,本发明将所述取代树脂、fmoc
‑
cys(trt)
‑
oh、偶联试剂和偶联反应溶剂混合进行第i偶联反应,得到第i偶联树脂。
[0089]
本发明优选对所述取代树脂进行前处理,在本发明中,所述前处理优选包括依次进行:溶胀、脱除fomc保护基、固液分离和洗涤;在本发明中,所述溶胀优选为:将所述起始树脂置于有机溶剂中进行溶胀,得到溶胀取代树脂。在本发明中,所述溶胀用有机溶剂优选为n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺或二氯甲烷,更优选为n,n
‑
二甲基甲酰胺。在本发明中,所述取代树脂的质量和有机溶剂的体积比优选为(7~12)g:(70~100)ml,更优选为(7.5~11)g:(75~90)ml。在本发明中,所述溶胀的温度优选为室温;所述溶胀的时间优选为30~150min,更优选为50~120min。溶胀结束后,本发明优选对溶胀后的溶胀体系进行固
液分离得到所述溶胀取代树脂,在本发明中,所述固液分离优选为过滤。在本发明中,所述溶胀中和掉初始树脂中多余的酸,通过溶胀使得树脂膨胀开,反应位点舒展,有利于初始树脂偶联氨基酸。
[0090]
本发明优选将所述溶胀取代树脂进行脱除fomc保护基,得到脱保护取代树脂,在本发明中,所述脱除fomc保护基优选为向所述溶胀取代树脂中加入fmoc脱保护试剂进行脱保护反应。在本发明中,所述fmoc脱保护试剂优选包括pip、dbu、tea或dblk,更优选为dblk;在本发明中,所述fmoc脱保护试剂优选以fmoc脱保护试剂溶液形式使用,所述fmoc脱保护试剂溶液的体积百分比优选为15~25%,更优选为20%。在本发明中,所述fmoc脱保护试剂溶液中的溶剂优选为n,n
‑
二甲基甲酰胺或n,n
‑
二甲基乙酰胺。在本发明中,所述取代树脂的质量和fmoc脱保护试剂溶液的体积比优选为(7~12)g:(100~150)ml,更优选为(7.5~11)g:(95~145)ml。在本发明中,所述脱保护反应的温度优选为室温,所述脱除fomc保护基的反应时间优选为25~40min,更优选为30min。本发明优选对所述脱除fomc保护基后的脱保护体系进行固液分离,所述固液分离优选为过滤,本发明对所述过滤的具体实施过程没有特殊要求。
[0091]
本发明优选对固液分离的固体产品进行洗涤,在本发明中,所述洗涤用溶剂优选为dmf,所述洗涤的次数优选为5次,在本发明中,所述取代树脂的质量和洗涤剂的体积比优选为(7~12)g:(100~150)ml,更优选为(7.5~11)g:(95~145)ml。
[0092]
在本发明中,所述取代树脂和所述fmoc
‑
cys(trt)
‑
oh的物质的量之比优选为1:(2~4),更优选为1:(1.5~3),最优选为1:2。
[0093]
在本发明中,所述偶联试剂优选包括dic/hobt或dic/hoat,更优选包括dic/hobt。在本发明中,所述偶联试剂中的dic为缩合试剂组分,所述hoat或hobt为催化剂组分;在本发明中,所述dic/hobt中dic的体积和hobt的质量比优选为(1~2)ml:(13~14)g。在本发明中,所述dic/hoat中dic的体积和hoat的质量比优选为(1~2)ml:(13~14)g。
[0094]
在本发明中,所述fmoc
‑
cys(trt)
‑
oh和偶联试剂的摩尔比优选为1:(1~1.2)。
[0095]
在本发明中,所述偶联溶剂优选包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺二氯甲烷(dcm)或四氢呋喃(thf),更优选包括n,n
‑
二甲基甲酰胺。本发明对于所述溶剂的用量没有特殊限定,能够将偶联试剂溶解即可。
[0096]
本发明将所述取代树脂、fmoc
‑
cys(trt)
‑
oh、偶联试剂和偶联溶剂混合的顺序优选为:将fmoc
‑
cys(trt)
‑
oh、偶联试剂中的催化剂组分和偶联溶剂进行第一混合,得到第一混合液后,本发明优选将所述第一混合进行降温处理,在本发明中,所述降温处理的温度优选为0℃,所述降温处理的保温时间优选为15~30min;本发明降温处理后的第一混合液与偶联剂中的缩合剂组分进行第二混合,再与所述取代树脂进行第三混合。在本发明中,所述第二混合的温度优选为室温;所述第二混合的时间优选为5~15min,更优选为10min。在本发明中,所述混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度没有特殊限定,能够将原料混合均匀即可。
[0097]
在本发明中,所述第i偶联反应的温度优选为20~40℃,更优选为室温;所述第i偶联反应的时间优选为1.5~2h。
[0098]
所述第i偶联反应后,本发明优选将所述第i偶联反应体系进行后处理,所述后处理优选包括:依次将所述第i偶联反应液固液分离,将所得固体产品进行有机溶剂洗涤,得
到第i偶联树脂。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤。在本发明中,所述有机溶剂优选包括n,n
‑
二甲基甲酰胺或n,n
‑
二甲基乙酰胺;在本发明中,所述取代树脂的质量和有机溶剂的体积比为(7~12)g:(100~150)ml;所述有机溶剂洗涤的次数优选为2~6次,更优选为5次;所述有机溶剂洗涤的目的是洗去未参加反应的氨基酸和偶联试剂。本发明优选通过茚三酮检测液检测判定偶联反应终点,当茚三酮检测为阴性时,则偶联反应完全,得到第i偶联树脂。
[0099]
得到第i偶联树脂后,本发明按照所述第i偶联反应,在所述第i偶联树脂依次偶联剩余的肽树脂结构中所需剩余保护氨基酸,得到所述肽树脂,所述肽树脂结构中所需的剩余保护氨基酸依次为fmoc
‑
gly
‑
oh、fmoc
‑
thr(tbu)
‑
oh、fmoc
‑
cys(acm)
‑
oh、fmoc
‑
ala
‑
oh、fmoc
‑
pro
‑
oh和fmoc
‑
asn(trt)
‑
oh。
[0100]
在本发明中,所述每种剩余的保护氨基酸和和偶联试剂的摩尔比优选为1:(1~1.2)。
[0101]
在本发明中,所述第i偶联树脂和所述每种剩余的保护氨基酸的物质的量之比优选为1:(2~4)。
[0102]
在本发明中,在所述第i偶联树脂依次偶联剩余的保护氨基酸的每一次偶联反应的操作以及条件独立地的优选与前述第i偶联树脂制备过程的操作以及条件相同,在此不再一一赘述。
[0103]
本发明优选对所述肽树脂进行前处理,在本发明中,所述前处理优选包括依次进行:溶胀、脱除fomc保护基、固液分离和洗涤;在本发明中,对所述肽树脂进行前处理的步骤和参数优选与所述取代树脂的前处理相同,在此不再一一赘述。
[0104]
在本发明中,所述带保护基团的胱氨酸化合物的结构式如式vi所示:
[0105][0106]
在本发明中,所述式vi中,x1为dde、alloc、boc或mtt,优选为boc,所述r1为oall或odmab,优选为oall。
[0107]
在本发明中,所述带保护基团的胱氨酸化合物的制备方法优选包括以下步骤:将fmoc
‑
cys
‑
oh和2,2'
‑
二硫二吡啶混合进行亚磺酰化反应,得到亚磺酰化产品,将所述亚磺酰化产品和x1
‑
cys
‑
r1混合进行取代反应。在本发明中,所述x1为dde、alloc、boc或mtt,优选为boc,所述r1为oall或odmab,优选为oall,在本发明中,所述fmoc
‑
cys
‑
oh、2,2'
‑
二硫二吡啶和x1
‑
cys
‑
r1的物质量之比优选为1:1:1;在本发明中,所述亚磺酰化反应的温度优选为是室温,所述取代反应的温度优选为室温。
[0108]
在本发明中,所述肽树脂和带保护基团的胱氨酸化合物的物质的量之比为1:(2~4)。在本发明中,所述偶联试剂优选包括dic/hobt或dic/hoat,更优选包括dic/hobt。在本发明中,所述偶联试剂中的dic为缩合试剂组分,所述hoat或hobt为催化剂组分;在本发明中,所述dic/hobt中dic的体积和hobt的质量比优选为(1~2)ml:(13~14)g。在本发明中,所述dic/hoat中dic的体积和hoat的质量比优选为(1~2)ml:(13~14)g。
[0109]
在本发明中,所述带保护基团的胱氨酸化合物和偶联试剂的摩尔比优选为1:(2~4)。
[0110]
在本发明中,所述偶联溶剂优选包括n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺二氯甲烷(dcm)或四氢呋喃(thf),更优选包括n,n
‑
二甲基甲酰胺。本发明对于所述溶剂的用量没有特殊限定,能够将偶联试剂溶解即可。
[0111]
在本发明中,本发明将所述肽树脂、带保护基团的胱氨酸化合物、偶联试剂和偶联溶剂混合的顺序优选为:将带保护基团的胱氨酸化合物、偶联试剂中的催化剂组分和偶联溶剂进行第一混合,得到第一混合液后,本发明优选将所述第一混合进行降温处理,在本发明中,所述降温处理的温度优选为0℃,所述降温处理的保温时间优选为15~30min;本发明降温处理后的第一混合液与偶联剂中的缩合剂组分进行第二混合,再与所述肽树脂进行第三混合。在本发明中,所述第二混合的温度优选为室温;所述第二混合的时间优选为5~15min,更优选为10min。在本发明中,所述混合的方式优选为搅拌混合,本发明对于所述搅拌混合的速度没有特殊限定,能够将原料混合均匀即可。
[0112]
在本发明中,所述第一偶联反应的温度优选为20~40℃,更优选为室温;所述第一偶联反应的时间优选为1.5~2h。
[0113]
所述第一偶联反应后,本发明优选将所述第一偶联反应体系进行后处理,得到所述第一中间体,在本发明中,所述第一偶联反应体系的后处理优选与所述第i偶联反应体系进行后处理时的步骤和参数相同,在此不再一一赘述。
[0114]
得到第一中间体后,本发明按照所述第一述偶联反应,在所述第一中间体依次偶联剩余保护氨基酸,得到所述第二中间体,所述剩余保护氨基酸依次为fmoc
‑
cys(trt)
‑
oh、fmoc
‑
tyr(tbu)
‑
oh、fmoc
‑
glu(otbu)
‑
oh和fmoc
‑
cys(acm)
‑
oh。
[0115]
所述第二中间体的结构如式ii所示:
[0116][0117]
在本发明中,在所述第一中间体依次偶联剩余保护氨基酸的每一次偶联反应的操作以及条件独立地的优选与前述第一偶联树脂制备过程的操作以及条件相同,在此不再一一赘述。
[0118]
得到第二中间体后,本发明将所述第二中间、脱保护试剂和催化剂混合脱除保护基团r1得到第三中间体,所述第三中间体的结构如式iii所示:
[0119][0120]
本发明优选对所述第二中间体进行前处理,在本发明中,所述前处理优选包括洗涤,在本发明中,所述洗涤优选为dcm洗涤,所述dcm洗涤的次数优选为3~5次,本发明对所述dcm的用量没有特殊要求。
[0121]
在本发明中,当所述保护基团r1为oall时,所述脱除保护试剂为醋酸、nmm和dcm的混合物,所述催化剂为钯催化剂;在本发明中,所述醋酸、nmm和dcm的摩尔比优选为0.5:1:8.5,所述第二中间体的质量和脱保护试剂的体积比优选为1g:10ml。在本发明中,所述催化剂优选为钯催化剂,更优选为四(三苯基膦)钯,在本发明中,所述催化剂的质量和脱保护试剂的体积比优选为1g:(100~120)ml。
[0122]
当所述保护基团r1为odmab时,所述脱除保护试剂为水合肼的dmf溶液,所述水合肼的dmf溶液中水合肼的质量百分比优选为2%。
[0123]
在本发明中,所述第二中间体、脱保护试剂和催化剂混合的顺序优选为:将所述第二中间体和脱保护试剂进行第一混合后加入所述催化剂进行第二混合。
[0124]
在本发明中,所述脱除保护基团r1的温度优选为室温,所述脱除保护基团r1的时间优选为3~5h,所述脱除保护基团r1的优选搅拌的条件下进行,本发明对所述搅拌的具体实施过程没有特殊要求。
[0125]
本发明优选对所述脱除保护基团r1的体系进行后处理,在本发明中,所述后处理优选为洗涤,所述洗涤优选为n,n
‑
二乙基二硫代氨基甲酸钠的dmf溶液和dipea的dmf溶液的混合溶液洗涤3~5次,所述n,n
‑
二乙基二硫代氨基甲酸钠的dmf溶液和dipea的dmf溶液的体积比优选为1:1,所述n,n
‑
二乙基二硫代氨基甲酸钠的dmf溶液中n,n
‑
二乙基二硫代氨基甲酸钠的体积百分比优选为5%,所述dipea的dmf溶液中dipea的体积百分比优选为1%,本发明使用n,n
‑
二乙基二硫代氨基甲酸钠的dmf溶液和dipea的dmf溶液的混合溶液洗涤时,每次洗涤的时间优选为10min;固液分离后采用dmf洗涤3~5次,本发明对所述dmf的用量没有特殊要求。
[0126]
得到所述第三中间体后,本发明按照上述第一偶联反应,所述第三中间体发生分子内偶联反应,得到第四中间体,所述第四中间体的结构如式iv所示:
[0127][0128]
在本发明中,在所述第三中间体进行分子内偶联反应的操作以及条件独立地的优选与前述第一偶联树脂制备过程的操作以及条件相同,在此不再一一赘述。
[0129]
本发明通过分子内偶联反应得到第1对二硫键。
[0130]
得到第四中间体后,本发明将所述第四中间体在酸性裂解液中进行裂解,得到第五中间体,所述第五中间体的结构如式v所示:
[0131][0132]
在本发明中,所述酸性裂解液优选为tfa和dmso的混合溶液,所述tfa和dmso的混合溶液中,所述dmso的体积浓度优选为5~30%,更优选为10~25%。
[0133]
在本发明中,所述第四中间体的质量和酸性裂解液的体积比优选为(10~13)g:150ml,更优选为(11~12)g:150ml。
[0134]
本发明优选将所述酸性裂解液进行冷冻处理再进行裂解,所述冷冻处理的温度优选为
‑
25~
‑
20℃,所述冷冻处理的时间优选为30min。本发明优选向经过冷冻处理的酸性裂解中加入所述第四中间产物进行裂解。
[0135]
在本发明中,所述裂解的温度优选为为20~40℃,所述裂解的时间优选为1.5~3h。在本发明中,所述裂解在搅拌条件下进行,本发明对所述搅拌的具体实施过程没有特殊要求。
[0136]
本发明通过裂解得到第2对二硫键。
[0137]
本发明优选对裂解后的体系进行过后处理,得到第五中间体,在本发明中,所述后处理优选包括依次进行固液分离、洗涤固体产物、收集滤液和洗涤液、加入反溶剂析出固体产品、固液分离得到固体产品、洗涤和干燥;本发明对所述固液分离的具体实施过程没有特
殊要求,本发明通过固液分离得到滤液和树脂,本发明优选通过tfa洗涤树脂2次并收集洗涤液;本发明收集滤液和洗涤液后加入反溶剂,析出固体产品,在本发明中,所述反溶剂优选为乙醚,所述乙醚的温度优选为0~5℃,本发明对所述反溶剂的加入量没有特殊要求,加入至无固体产品析出即可,本发明优选通过固液分离得到固体产品,在本发明中,所述固液分离优选为离心,得到固体产品后,本发明对固体产品进行干燥,在本发明中,所述干燥优选为真空干燥,所述干燥的温度优选为20~35℃,更优选为25~30℃;本发明将所述固体产品干燥至失重<3%停止干燥。
[0138]
得到第五中间体后,本发明将所述第五中间体、酸性溶剂和碘氧化试剂混合进行氧化,得到所述利那洛肽。
[0139]
在本发明中,所述碘氧化试剂优选为碘的甲醇溶液或碘的乙腈溶液,所述碘氧化试剂中碘的质量浓度优选为30~35g/l,更优选为32~34g/l。
[0140]
在本发明中,所述酸性溶剂优选为醋酸水溶液,所述醋酸水溶液中醋酸的体积百分比优选为10%。
[0141]
在本发明中,所述第五中间体、酸性溶剂和碘氧化试剂混合得到的混合液中,所述第五中间体的质量浓度优选为1~10mg/ml,更优选为2~8mg/ml。
[0142]
在本发明中,所述第五中间体、酸性溶剂和碘氧化试剂混合的顺序优选为:将所述第五中间体和酸性溶剂进行第一混合后与所述碘氧化试剂进行第二混合。
[0143]
在本发明中,所述氧化的温度优选为5~45℃,更优选为20~40℃;所述氧化反应的时间优选为10~20min,更优选为12~18min,所示氧化反应优选在搅拌条件下进行,本发明对所述搅拌的具体实施过程没有特殊要求。
[0144]
本发明通过氧化得到第3对二硫键。
[0145]
本发明优选对所述氧化反应后的体系进行后处理,得到所述利那洛肽,在本发明中,所述后处理优选包括:依次进行:除去过量的碘氧化试剂得到利那洛肽粗品溶液,对所述利那洛肽粗品溶液进行纯化、转盐和干燥,得到所述利那洛肽;本发明优选向所述氧化反应后的体系中加入vc水溶液中和过量碘氧化试剂中的碘单质,本发明对所述vc水溶液的质量含量没有特殊要求,在本发明中,所述vc的用量以氧化反应后的体系的颜色消失为准(即碘单质无剩余)。在本发明中,所述纯化优选为色谱柱纯化,
[0146]
在本发明中,所述色谱柱纯化优选为半制备液相色谱纯化,所述半制备液相色谱纯化的条件包括:色谱柱优选为反相c18的dac200;流动相a优选为0.05~0.2v/v%tfa水溶液,更优选为0.1v/v%tfa水溶液;流动相b优选为乙腈;流动相a和流动相b的流速优选独立地为500~1000ml/min,更优选为700~800ml/min;检测波长优选为220nm;洗脱方式优选为一次上样,梯度洗脱;所述梯度洗脱的具体程序为:0~1h,流动相a的体积百分含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0147]
所述色谱柱纯化后,本发明优选还包括将所述色谱柱纯化得到的目标峰(保留时间为28.5~36.5min)洗脱液转盐后冻干,得到醋酸利那洛肽。在本发明中,所述转盐优选为半制备液相色谱转盐,所述半制备液相色谱转盐的条件包括:色谱柱优选为反相c18的dac200;流动相a优选为0.1v/v%醋酸水溶液,流动相b优选为乙腈,流动相a和流动相b的流速优选独立地为500~1000ml/min,更优选为700~800ml/min;检测波长优选为220nm;洗脱方式优选为一次上样,梯度洗脱;所述梯度洗脱的具体程序为:0~1h,流动相a的体积百分
含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0148]
本发明手机洗脱液进行干燥,在本发明中,所述干燥的方式优选为冻干,在本发明中,所述冻干优选采用冻干机进行,所述冻干机的规格优选为0.5m2;所述冷冻干燥的时间为72h。
[0149]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0150]
实施例1
[0151]
称取200g取代度为1.1mmol/g的2
‑
cl trt
‑
cl树脂和202gfmoc
‑
tyr(tbu)
‑
oh,倒入20升反应釜中,加入3000ml的dcm,搅拌均匀使其溶解后,加入191ml的dipea,25℃水浴搅拌取代反应3小时,随后加入200ml的phlc级甲醇封闭30分钟;抽调反应液,用dmf洗涤2次、甲醇洗涤2次、dmc洗涤2次、甲醇洗涤2次;抽干滤液,将树脂取出进行真空干燥,直到干燥失重小于3%;得到fmoc
‑
tyr
‑
tyr(tbu)
‑
ctc resin284.51g,利用紫外分光光度计测定其取代度为:0.713mmol/,收率为83.68%。
[0152]
称取70.12g fmoc
‑
tyr
‑
tyr(tbu)
‑
ctc resin(50mmol)置于多肽反应釜中,加入700mldmf溶胀树脂2小时,抽掉溶剂,加入1000ml的dblk溶液进行脱保护30分钟,抽滤掉溶剂,每次加入1000mldmf洗涤树脂5次;称取58.5g fmoc
‑
cys(trt)
‑
oh和135.1g hobt,加入活化灌内用500ml dmf溶解,冰水浴冷却15分钟后加入15.7ml dic活化5分钟;将活化完的溶液倒入反应釜中,加入适量的dmf使其能够均匀搅拌,30℃反应2小时(茚三酮检测阴性);抽滤掉溶剂,每次加入1000ml dmf洗涤5次。同样的操作流程继续依次偶联以下氨基酸:fmoc
‑
gly
‑
oh(29.7g,100mmol)、fmoc
‑
thr(tbu)
‑
oh(39.7g,100mmol)、fmoc
‑
cys(acm)
‑
oh(58.5g,100mmol)、fmoc
‑
ala
‑
oh(31.1g,100mmol)、fmoc
‑
pro
‑
oh(33.7g,100mmol)、fmoc
‑
asn(trt)
‑
oh(59.7g,100mmol)、fmoc
‑
cys(boc
‑
cys
‑
oall)
‑
oh(60.5g,100mmol)、fmoc
‑
cys(trt)
‑
oh(58.5g,100mmol)、fmoc
‑
tyr(tbu)
‑
oh(45.9g,100mmol)、fmoc
‑
glu(otbu)
‑
oh(42.5g,100mmol)和fmoc
‑
cys(acm)
‑
oh(41.4g,100mmol),最终得到第二中间体,结构式如下:
[0153][0154]
取第二中间体1,每次加入1000ml dcm洗涤树脂3次,加入醋酸:nmm:dcm体积比为0.5:1:8.5的溶液1000ml,加入四(三苯基膦)钯10g,搅拌反应3小时,抽掉溶剂,每次加入1000ml的体积浓度为5%n,n
‑
二乙基二硫代氨基甲酸钠/dmf溶液和体积浓度为1%dipea/dmf的混合溶液(体积比为1:1)洗涤树脂5次,每次10分钟,再每次加入1000ml的dmf洗涤树脂3次,然后加入135.1g hobt、15.7ml dic,适量600mldmf搅拌反应3小时(茚三酮检测呈阴性),抽滤掉反应液,每次加入1000ml的dmf洗涤树脂5次,每次加入1000ml的甲醇洗涤树脂2次,每次加入1000ml的dcm洗涤树脂2次,每次加入1000ml的甲醇洗涤树脂2次,抽干滤液,将树脂取出进行真空干燥,干燥温度**℃,直到干燥失重小于3%,得到第四中间体,201.23g,合成收率:93.81%,结构如下:
[0155][0156]
取第四中间体100.62g,配置dmso的tfa溶液((dmso和tfa的体积比为2.5:7.5)1500ml,置于
‑
20℃冷却30分钟,将第四中间体缓慢倒入dmso的tfa溶液中,一边搅拌一边倒入肽树脂,将肽树脂全部倒入裂解液中,25℃搅拌2.5小时,过滤取滤液,每次加入150ml tfa洗涤树脂2次,合并滤液,用10升冰乙醚沉淀,离心机离心取沉淀,并用乙醚洗涤沉淀3次,真空干燥,直到干燥失重小于3%,得到第五中间体,41.2g,纯度81.25%,收率107.44%,结构如下:
[0157][0158]
称取第五中间体20.6g,溶于2.1升醋酸水溶液中(体积百分比10%醋酸),加入碘的甲醇溶液(碘的质量浓度为30g/l)100ml,室温搅拌反应30分钟,加入适量vc水溶液中和过剩的碘(溶液中颜色消失为止),制得利那洛肽粗品溶液,粗品纯度:80.56%,其结构式如下
[0159][0160]
将利那洛肽粗品溶液,采用奥诺的半制备液相色谱仪进行纯化,收集目标峰(保留时间为28.5~36.5min)的洗脱液,采用0.5m2冻干机冷冻干燥72小时,得到醋酸利那洛肽精品:9.57g,纯度99.61%:总收率:50.17%。
[0161]
其中,hplc纯化的条件包括:色谱柱为反相c18的dac200;检测波长为220nm;流动相a为0.1v/v%tfa水溶液;流动相b为乙腈;流动相a和流动相b的流速均为800ml/min;洗脱方式为梯度洗脱;所述梯度系统的具体程序为:0~1h,流动相a的体积百分含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0162]
hplc转盐的条件包括:色谱柱为反相c18的dac200;检测波长为220nm;流动相a为0.1v/v%醋酸水溶液;流动相b为乙腈;流动相a和流动相b的流速均为800ml/min;洗脱方式为梯度洗脱;所述梯度系统的具体程序为:0~1h,流动相a的体积百分含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0163]
本实施例的hplc检测结果如图1和表1所示。
[0164]
表1实施例1制备的醋酸利那洛肽的hplc检测结果
[0165]
保留时间(min)峰面积(mv
·
s)峰面积占比(%)峰高度(mv)28.5~30.5909918616.681930.5~32.51870824634.2174732.5~34.51714505931.4158634.5~36.5850034615.5785
[0166]
由图1和表1可知,本发明的工艺得到的粗品纯度高,纯化难度低,杂质控制良好,纯化收率高。
[0167]
实施例2
[0168]
称取200g取代度为0.66mmol/g的2
‑
cl trt
‑
cl树脂和121gfmoc
‑
tyr(tbu)
‑
oh,倒入20升反应釜中,加入3000ml的dcm,搅拌均匀使其溶解后,加入115ml的dipea,25℃水浴搅拌取代反应3小时,随后加入200ml的phlc级甲醇封闭30分钟;抽调反应液,用dmf洗涤2次、甲醇洗涤2次、dmc洗涤2次、甲醇洗涤2次;抽干滤液,将树脂取出进行真空干燥,直到干燥失重小于3%;得到fmoc
‑
tyr
‑
tyr(tbu)
‑
ctc resin254.87g,利用紫外分光光度计测定其取代度为:0.459mmol/,收率为90.56%。
[0169]
称取108.9g fmoc
‑
tyr
‑
tyr(tbu)
‑
ctc resin(50mmol)置于多肽反应釜中,加入1500mldmf溶胀树脂2小时,抽掉溶剂,加入1500ml的dblk溶液进行脱保护30分钟,抽滤掉溶剂,每次加入1500mldmf洗涤树脂5次;称取58.5g fmoc
‑
cys(trt)
‑
oh和135.1g hobt,加入活化灌内用500ml dmf溶解,冰水浴冷却15分钟后加入15.7ml dic活化5分钟;将活化完的溶液倒入反应釜中,加入适量的dmf使其能够均匀搅拌,30℃反应2小时(茚三酮检测阴性);抽滤掉溶剂,每次加入1000ml dmf洗涤5次。同样的操作流程继续依次偶联以下氨基酸:fmoc
‑
gly
‑
oh(29.7g,100mmol)、fmoc
‑
thr(tbu)
‑
oh(39.7g,100mmol)、fmoc
‑
cys(acm)
‑
oh(41.4g,100mmol)、fmoc
‑
ala
‑
oh(31.1g,100mmol)、fmoc
‑
pro
‑
oh(33.7g,100mmol)、fmoc
‑
asn(trt)
‑
oh(59.7g,100mmol)、fmoc
‑
cys(boc
‑
cys
‑
oall)
‑
oh(60.5g,100mmol)、fmoc
‑
cys(trt)
‑
oh(58.5g,100mmol)、fmoc
‑
tyr(tbu)
‑
oh(45.9g,100mmol)、fmoc
‑
glu(otbu)
‑
oh(42.5g,100mmol)和fmoc
‑
cys(acm)
‑
oh(41.4g,100mmol),最终得到第二中间体,结构式如下:
[0170][0171]
取第二中间体,每次加入1500ml dcm洗涤树脂3次,加入醋酸:nmm:dcm体积比为0.5:1:8.5的溶液1500ml,加入四(三苯基膦)钯10g,搅拌反应3小时,抽掉溶剂,每次加入1500ml的体积浓度为5%n,n
‑
二乙基二硫代氨基甲酸钠/dmf溶液和体积浓度为1%dipea/dmf的混合溶液(体积比为1:1)洗涤树脂5次,每次10分钟,再每次加入1000ml的dmf洗涤树脂3次,然后加入135.1g hobt、15.7ml dic,600mldmf搅拌反应3小时(茚三酮检测呈阴性),抽滤掉反应液,每次加入1500ml的dmf洗涤树脂5次,每次加入1500ml的甲醇洗涤树脂2次,每次加入1500ml的dcm洗涤树脂2次,每次加入1500ml的甲醇洗涤树脂2次,抽干滤液,将树脂取出进行真空干燥,干燥温度40℃,直到干燥失重小于3%,得到第四中间体,241.68g,合成收率:95.01%,结构如下:
[0172][0173]
取第四中间体120.84g,配置dmso的tfa溶液(dmso和tfa的体积比为2.5:7.5)1500ml,置于
‑
20℃冷却30分钟,将第四中间体缓慢倒入dmso的tfa溶液中,一边搅拌一边倒入肽树脂,将肽树脂全部倒入裂解液中,25℃搅拌2.5小时,过滤取滤液,每次加入150ml tfa洗涤树脂2次,合并滤液,用10升冰乙醚沉淀,离心机离心取沉淀,并用乙醚洗涤沉淀3次,真空干燥,直到干燥失重小于3%,得到第五中间体,42.5g,纯度83.02%,收率110.94%,结构如下:
[0174][0175]
称取第五中间体23.2g,溶于2.1升醋酸水溶液中(体积百分比10%醋酸),加入碘的甲醇溶液(电的质量浓度为30g/l)100ml,室温搅拌反应30分钟,加入适量vc水溶液中和过剩的碘(溶液中颜色消失为止),制得利那洛肽粗品溶液,粗品纯度:80.56%,其结构式如下
[0176][0177]
将利那洛肽粗品溶液,采用奥诺的半制备液相色谱仪进行纯化,收集目标峰(保留时间为28.3
‑
36.3min)的洗脱液,采用0.5m2冻干机冷冻干燥72小时,得到醋酸利那洛肽精品:12.04g,纯度99.67%:总收率:51.89%。
[0178]
其中,hplc纯化的条件包括:色谱柱为反相c18的dac200;检测波长为220nm;流动相a为0.1v/v%tfa水溶液;流动相b为乙腈;流动相a和流动相b的流速均为800ml/min;洗脱方式为梯度洗脱;所述梯度系统的具体程序为:0~1h,流动相a的体积百分含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0179]
hplc转盐的条件包括:色谱柱为反相c18的dac200;检测波长为220nm;流动相a为0.1v/v%醋酸水溶液;流动相b为乙腈;流动相a和流动相b的流速均为800ml/min;洗脱方式为梯度洗脱;所述梯度系统的具体程序为:0~1h,流动相a的体积百分含量由20%匀速增加至60%,流动相b的体积百分含量由80%匀速降低至40%。
[0180]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。