1.本发明属于有机合成,具体涉及一种制备芳基异硫脲的方法。
背景技术:
2.s
‑
芳基异硫脲化合物在农业、化学、医药领域具有广泛的应用[“phenoxyphenylthioureas”: drabek j, b
ç
ger m, fr2465720a1, 1980, and 4.939.257, 1988。nicholson a, perry j d, james a l, stanforth s p, et al. int. j. antimicrob. agents2012, 39, 27
‑
32],吸引了众多化学家和医药学家浓厚的兴趣。农业上,异硫脲类衍生物可以用做除草剂[kogan m,dinizo s e,fancher l w, br8602373a. 1987]、杀虫剂[drabek j, boeger m, ehrenfreund j, et al. rec. adv. chem. insect control ii. 1990, 170
‑
183]、杀螨剂[pascual a, rindlisbacher a. pest manage. sci. 1994, 42, 253
‑
263]等。化学合成上,异硫脲结构的化合物可被用作催化剂[taylor j e, bull s d, williams j. cheminform abstract: amidines, isothioureas, and guanidines as nucleophilic catalysts. chem. soc. rev. 2012, 41, 2109
‑
2121]和过渡金属催化的配体。在医药方面应用更广,拥有很好的抗病毒、抗组胺活性。因此,越来越多的异硫脲药物分子被合成与设计,表现出非常有意义的生物学作用,有效促进了医药学领域的发展,已经有hiv
‑
1抑制剂、抗感染剂、中枢神经剂和缬氨酸蛋白抑制剂等方面的研究。
[0003]
自进入21世纪以来,金属催化偶联反应迎来一股热潮,chan
‑
lam反应已成为构建c
‑
s键的一种有效、实用的替代方法。dong课题组对异硫脲的合成及其应用方面有着长期的研究兴趣,近些年报道了一系列通过金属催化剂将硫脲转化为s
‑
芳基异硫脲的简易方法。2018年,以cu(oac)2·
h2o为催化剂,联吡啶为配体,合成了理想的s
‑
芳基异硫脲,收率基本上都有90%[ liu x, zhang s b, zhu h, et al. an efficient chan
‑
lam s
‑
arylation of arylthioureas with aryl boronic acids. eur. j. org. chem. 2018, 4483
‑
4489],各类官能团取代的芳基硫脲和芳基硼酸都很耐受。而后,他们继续用廉价的金属铜做催化剂,在无需配体参与下,将碘代苯与硫脲偶联生成s
‑
芳基异硫脲[zhu h, liu x, chang c z, et al. copper
‑
catalyzed c
‑
s crosscoupling reaction: s
‑
arylation of arylthioureas. synthesis. 2017, 49, 5211
‑
5216],原料碘代苯的来源比苯硼酸更广泛。为提高在工业生产上的适用性,金属催化的异硫脲合成方案一直被优化、改进。maes课题组报道了使用cui催化苯胺、磺酸酯和异氰化物三组分合成s
‑
芳基化和s
‑
烷基化异硫脲的新方法[mampuys p, zhu y, vlaar t, et al. angew. chem. int. ed. 2014, 53, 12849
‑
12854]。在铜金属催化下,作者首次实现了将异氰化物插入到磺酸酯中形成异硫氰酸酯硫鎓中间体,随后与苯胺反应生成异硫脲。
[0004]
s
‑
芳基异硫脲结构存在于许多化学分子中,广泛应用于功能材料和药物领域,吸引了科学家们浓厚的兴趣。除了反应原料需要简单易得之外,反应温度、反应时间尽可能温和,也是有机合成追求的方向。
技术实现要素:
[0005]
本发明公开了一种制备芳基异硫脲的方法,能在温和、经济且简便的条件下与硫脲发生亲核反应生成s
‑
芳基异硫脲,尤其是,反应制备产物的时间小于5小时,既节约能源,又避免长时间反应可能带来的反应不稳定问题,这是非常可取的芳基c
‑
s键形成方案。
[0006]
本发明采用如下技术方案:一种制备芳基异硫脲的方法,将氢化物悬于溶剂中,然后依次加入硫脲、邻二碘苯,接着反应,制备芳基异硫脲。
[0007]
本发明中,硫脲的化学结构式如下:邻二碘苯的化学结构式如下:本发明芳基异硫脲的化学结构式如下:上述结构式中,r1、r2、r3独立的选自氢、吸电子基团、给电子基团或者保护基团,比如卤素、氰基、烯基、取代或未取代的烷基、烷氧基、取代或未取代的氨基、取代或未取代的苯基、取代或未取代的杂环基;进一步的,取代指基团上的一个或多个氢原子被取代基取代;取代的烷基、取代的氨基中取代基独立的选自卤素、oh、nh2、cn、未取代或卤代的c1~c8烷基、未取代或卤代的c3~c8环烷基、未取代或卤代的c1~c8烷氧基、未取代或卤代的c2~c6烯基、未取代或卤代的c2~c6炔基、未取代或卤代的c2~c6酰基、未取代或卤代的4~8元饱和杂环或碳环中的一种或几种;其中,所述的杂环包含n、o、s中的一种或几种。或者r1、r2组成环状基团,具体的,r1、r2与连接r1、r2的n组成环状基团,该环状基团可以为单环结构,也可以为多环结构。
[0008]
本发明公开的硫脲与邻二碘苯的反应在金属氢化物存在下、溶剂中进行,无需其他物质,室温下反应1~4小时,得到产物芳基异硫脲为单一产物。
[0009]
本发明中,金属氢化物为氢化钠、氢化钾、氢化钙、氢化锂等;金属氢化物的用量为硫脲摩尔量的3~6倍。进一步的,邻二碘苯的用量为硫脲摩尔量的1~3倍。
[0010]
本发明中,溶剂为二甲基乙酰胺dma、四氢呋喃thf、乙腈ch3cn、乙二醇二甲醚dme、甲苯toluene中的一种或几种,优选为thf和dma,两者体积比优选为(4~7)∶1。
[0011]
金属催化的方法虽然适用于异硫脲化合物的合成,但也存在需要高温,反应时间长,催化剂负载量大,试剂价格昂贵,且容易造成金属废弃物污染的缺点。因此,开发无金属催化、原料廉价、非空气敏感的反应系统制备s
‑
芳基异硫脲是非常需要的。近几年,化学家们开始尝试直接使用n位取代的咪唑与二硫化物发生亲核取代反应,无需金属催化剂方便的生成s
‑
芳基化咪唑,但需要使用n
‑
buli进行脱质子化,反应需无水、无氧氛围,安全性差。本发明在nah作用下使用邻二碘苯与硫脲化合物进行亲核加成反应,实现了首次利用邻二碘苯直接与硫脲进行c
‑
s偶联生成s
‑
(2
‑
碘芳基)异硫脲,且邻位取代的二碘苯与硫脲反应
具有很好的区域选择性。该方案操作十分简便,无需金属催化,原料廉价易得,官能团耐受性好。为s
‑
芳基异硫脲砌块的药物合成提供了一种优秀方案,对未来的药物合成发展具有重大意义。
附图说明
[0012]
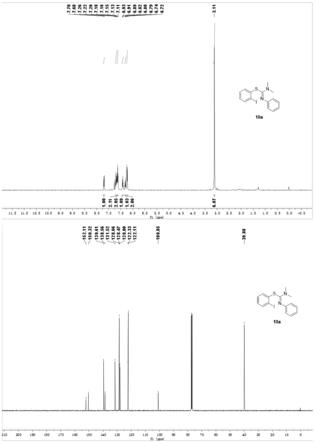
图1为化合物10a的核磁谱图;图2为化合物10n的x射线晶体学分析图。
具体实施方式
[0013]
本发明以硫脲与邻二碘苯为底物,在金属氢化物以及溶剂存在下,室温即可完成反应,高收率得到产物芳基异硫脲,无需其他物质,解决了现有技术需要金属催化剂、格式试剂等问题。
[0014]
本发明涉及的原料都是现有产品,可市购,也可根据现有方法制备。
[0015]
化合物核磁h谱由agilent 400 mhz与bruker 400 mhz仪器检测,c谱由bruker 400 mhz仪器检测,样品溶剂为氘代试剂(cdcl3或d6‑
dmso),均含有tms内标,核磁数据报告包括:化学位移,峰面积积分,偶合常数,峰型等。单晶检测使用x
‑
射线单晶衍射仪(d8 quest)。tlc薄层色谱板为烟台黄海化工厂生产,在254nm或365nm波长下可视化监测,显色剂有kmno4、碘、磷钼酸和二硝基苯肼,快速柱层析所用硅胶目数为200
‑
300目。所用试剂都为市售分析纯或化学纯,无特殊说明,直接使用。无水溶剂均为重蒸溶剂或市售干燥溶剂(百灵威)。
[0016]
除非另有说明,本发明采用本领域技术范围内的常规方法,如质谱、nmr、ir和uv/vis光谱法。除非提出具体定义,否则本文在分析化学、有机合成化学的有关描述中采用的术语是本领域已知的。可在化学合成、化学分析中使用标准技术。在本说明书中,可由本领域技术人员选择基团及其取代基以提供稳定的结构部分和化合物。当通过从左向右书写的常规化学式描述取代基时,该取代基也同样包括从右向左书写结构式时所得到的在化学上等同的取代基。举例而言,
‑
ch2o
‑
等同于
‑
och2‑
。在本文中定义的某些化学基团前面通过简化符号来表示该基团中存在的碳原子总数。例如,c1
‑
6烷基是指具有总共1至6个碳原子的如下文所定义的烷基。简化符号中的碳原子总数不包括可能存在于所述基团的取代基中的碳。
[0017]
在本发明中,卤素是指氟、氯、溴或碘;羟基是指
‑
oh基团;羟基烷基是指被羟基(
‑
oh)取代的烷基;羰基是指
‑
c(=o)
‑
基团;硝基是指
‑
no2;氰基是指
‑
cn;氨基是指
‑
nh2;羧基是指
‑
cooh。
[0018]
实施例一 芳基异硫脲的制备将nah(1.2 mmol, 4.0 equiv)称量于反应瓶中,悬于0.8 ml无水thf中常规搅拌,在搅拌过程中滴加硫脲9(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),继续在室温下搅拌,tlc监测反应完成。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到产品芳基异硫脲10。
[0019]
不同反应底物硫脲9以及得到的对应产物芳基异硫脲10如下:上述收率为分离收率,标注的时间为tlc监测反应完成的时间,上标b表示氢化钠用量为5当量,上标c表示氢化钠用量为4.5当量。
[0020]
上述部分产物数据表征如下:
1
h nmr (400 mhz, cdcl3) δ 7.69 (d, j = 7.8 hz, 1h), 7.19 (dd, j = 15.2, 7.2 hz, 2h), 7.13 (t, j = 7.5 hz, 2h), 6.91 (t, j = 7.3 hz, 1h), 6.80 (t, j = 7.0 hz, 1h), 6.73 (d, j = 7.6 hz, 2h), 3.11 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 152.11, 150.32, 139.61, 138.56, 131.52, 128.66, 128.47, 128.00, 122.33, 122.11, 100.85, 39.88。
[0021]1h nmr (400 mhz, cdcl3) δ 7.69 (d, j = 7.8 hz, 1h), 7.14 (d, j = 3.8 hz, 2h), 7.02 (d, j = 8.3 hz, 2h), 6.82 (dd, j = 7.7, 4.1 hz, 1h), 6.60 (d, j = 8.3 hz, 2h), 3.14 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 152.56, 148.87, 139.69, 137.92, 131.90, 128.65, 128.36, 128.27, 127.33, 123.33, 101.29, 39.97。
[0022]1h nmr (400 mhz, cdcl3) δ 7.63 (dd, j = 7.9, 1.2 hz, 1h), 7.16
ꢀ–ꢀ
7.10 (m, 2h), 7.06 (m, 2h), 6.87
ꢀ–ꢀ
6.80 (m, 2h), 6.79
ꢀ–ꢀ
6.71 (m, 1h), 3.21 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 153.04, 150.45, 139.66, 137.13, 132.83, 130.52 (d, j = 31.3 hz), 128.75, 128.54 (d, j = 8.1 hz), 125.66, 125.10, 122.95, 119.03 (q, j = 11.1 hz), 118.39 (q, j = 12.1 hz), 102.03, 40.07. 19
f nmr (377 mhz, cdcl3) δ
ꢀ‑
62.49。
[0023]1h nmr (400 mhz, cdcl3) δ 7.64 (dd, j = 7.9, 1.3 hz, 1h), 7.10 (dtd, j = 9.1, 7.9, 1.5 hz, 2h), 6.87 (dd, j = 8.8, 0.7 hz, 2h), 6.78 (m, 1h), 6.65
ꢀ–ꢀ
6.57 (m, 2h), 3.19 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 152.82, 148.86, 144.10, 144.08, 139.69, 137.45, 132.62, 128.57, 128.38, 122.76, 121.98, 121.24, 119.44, 101.98, 40.04。
[0024]1h nmr (400 mhz, cdcl3) δ 8.07
ꢀ–ꢀ
8.03 (m, 1h), 7.98 (t, j = 1.6 hz, 1h), 7.67
ꢀ–ꢀ
7.62 (m, 1h), 7.11 (dt, j = 5.5, 3.4 hz, 2h), 6.92 (dd, j = 3.4, 1.3 hz, 2h), 6.78 (m, 1h), 3.19 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 153.51, 146.50, 143.38, 142.58, 139.74, 137.21, 132.33, 129.08, 128.72, 128.56, 123.09, 101.57, 40.04。
[0025]1h nmr (400 mhz, cdcl3) δ 8.00 (d, j = 7.6 hz, 1h), 7.69
ꢀ–ꢀ
7.63 (m, 1h), 7.50
ꢀ–ꢀ
7.44 (m, 1h), 7.35 (m, 3h), 7.25 (d, j = 7.0 hz, 1h), 7.04 (dd, j = 7.8, 1.2 hz, 1h), 6.87 (dd, j = 11.0, 4.2 hz, 1h), 6.81 (d, j = 7.2 hz, 1h), 6.58
ꢀ–ꢀ
6.48 (m, 1h), 3.25 (s, 6h). 13
c nmr (101 mhz, cdcl3) δ 152.51, 146.83, 139.38, 137.41, 134.08, 132.43, 128.23, 128.17, 127.84, 127.61, 125.89, 125.67, 124.64, 124.07, 122.29, 116.42, 102.20, 40.14。
[0026]1h nmr (400 mhz, cdcl3) δ 7.68 (dd, j = 7.9, 1.0 hz, 1h), 7.22 (dd, j = 7.9, 1.4 hz, 1h), 7.12 (m, 3h), 6.87 (t, j = 7.4 hz, 1h), 6.79 (td, j = 7.8, 1.5 hz, 1h), 6.73 (d, j = 7.4 hz, 2h), 3.57 (t, j = 6.6 hz, 4h), 1.93
ꢀ–ꢀ
1.87 (m, 4h). 13
c nmr (101 mhz, cdcl3) δ 150.32, 148.21, 139.56, 138.38, 131.28, 128.67, 128.40, 127.92, 122.40, 122.16, 101.05, 49.23, 25.50。
141.75, 139.78, 139.12, 139.09, 138.75, 133.23, 128.76 (d, j = 8.1 hz), 128.63, 128.55 (d, j = 1.0 hz), 122.92, 121.92, 115.58 (d, j = 21.2 hz), 107.91, 105.71, 102.63, 101.18, 98.16, 68.80, 51.77, 48.68, 44.28, 41.74, 33.63. 19
f nmr (377 mhz, cdcl3) δ
ꢀ‑
116.05。
[0033]1h nmr (400 mhz, cdcl3) δ 8.39 (d, j = 8.2 hz, 1h), 7.79 (d, j = 7.6 hz, 1h), 7.67 (d, j = 7.9 hz, 1h), 7.53
ꢀ–ꢀ
7.46 (m, 2h), 7.42 (d, j = 8.2 hz, 1h), 7.29 (d, j = 7.9 hz, 1h), 7.23 (d, j = 5.0 hz, 1h), 7.20 (d, j = 7.8 hz, 1h), 7.16
ꢀ–ꢀ
7.07 (m, 4h), 6.99
ꢀ–ꢀ
6.94 (m, 1h), 6.90 (t, j = 7.3 hz, 1h), 6.85 (d, j = 7.7 hz, 1h), 6.78 (t, j = 7.5 hz, 1h), 6.69 (d, j = 7.8 hz, 2h), 5.73 (dd, j = 8.0, 4.8 hz, 1h), 3.93
ꢀ–ꢀ
3.73 (m, 2h), 3.13 (s, 3h), 2.63
ꢀ–ꢀ
2.38 (m, 2h). 13
c nmr (101 mhz, cdcl3) δ 153.30, 151.35, 150.08, 144.82, 139.57, 138.32, 134.71, 131.72, 128.62, 128.42, 128.10, 127.60, 126.78, 126.47, 126.19, 125.78, 125.44, 124.99, 124.85, 122.26, 122.00, 120.89, 107.09, 101.17, 74.48, 49.05, 38.41, 37.14。
[0034]1h nmr (400 mhz, cdcl3) δ 7.69 (dd, j = 7.9, 1.2 hz, 1h), 7.29 (dd, j = 7.9, 1.5 hz, 1h), 7.20
ꢀ–ꢀ
7.15 (m, 2h), 7.13 (s, 1h), 6.95 (t, j = 7.3 hz, 2h), 6.82 (m, 1h), 6.78
ꢀ–ꢀ
6.73 (m, 2h), 6.63 (d, j = 8.5 hz, 1h), 3.87 (d, j = 1.3 hz, 6h), 3.85 (s, 3h), 3.64
ꢀ–ꢀ
3.58 (m, 4h), 3.44 (s, 2h), 2.37
ꢀ–ꢀ
2.31 (m, 4h). 13
c nmr (101 mhz, cdcl3) δ 153.03, 152.74, 152.62, 149.89, 142.33, 139.57, 138.40, 132.49, 128.46, 128.43, 128.20, 125.08, 122.58, 121.79, 106.99, 101.54, 61.22, 60.82, 56.40, 56.01, 52.51, 47.94。
[0035]1h nmr (400 mhz, cdcl3) δ 7.68 (dd, j = 7.9, 0.8 hz, 1h), 7.44 (d, j = 8.7 hz, 2h), 7.40
ꢀ–ꢀ
7.33 (m, 4h), 7.30 (m, 1h), 7.21
ꢀ–ꢀ
7.12 (m, 2h), 7.09 (t, j = 7.8 hz, 2h), 6.97
ꢀ–ꢀ
6.86 (m, 3h), 6.83
ꢀ–ꢀ
6.77 (m, 1h), 6.60 (d, j = 7.3 hz, 2h), 5.24 (dd, j = 9.0, 4.0 hz, 1h), 3.73 (m, 2h), 3.12 (s, 3h), 2.35
ꢀ–ꢀ
2.15 (m, 2h). 13
c nmr (101 mhz, cdcl3) δ 160.60, 151.64, 150.15, 140.98, 139.72, 138.50, 131.84, 129.13, 128.73, 128.55, 128.22, 127.01 (q, j = 11.1 hz), 125.94, 125.89, 123.28 (d, j = 6.1 hz), 122.99, 122.46, 122.04, 116.04, 101.23, 78.41, 49.15, 38.26, 37.07. 19
f nmr (377 mhz, cdcl3) δ
ꢀ‑
61.52。
[0036]
上述底物硫脲9的结构式可根据产物结构式看出,如下:上述底物硫脲9的结构式可根据产物结构式看出,如下:r为苯基、取代苯基、杂环、萘基等;r1、r2如前文。
[0037]
具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:具体可如下:
本发明的反应底物可常规市购也可根据常规方法制备,比如在室温下,使用n2进行保护,将异硫氰酸酯(2.0 mmol)称量于两口瓶中,加入5 ml thf进行磁力搅拌。然后用注射器添加2m浓度的二甲胺
‑
thf溶液(1.1 equiv),继续在室温下搅拌反应,tlc监测反应进程。反应完成,将反应液转移到单口圆底瓶,直接旋干溶剂,然后用石油醚和乙酸乙酯混合溶剂打浆,抽滤,收集滤饼,得到纯净白色固体产品。
[0038]
在室温下,使用n2进行保护,将苯基异硫氰酸酯(2.0 mmol)称量于两口瓶中,加入5 ml etoh进行磁力搅拌。然后用注射器添加etoh溶解的仲胺化合物(1.1 equiv),继续在室温下搅拌反应,tlc监测反应进程。反应完成,反应液中有白色不溶固体析出,将反应液转移到单口圆底瓶,直接旋干溶剂,然后用石油醚和乙酸乙酯混合溶剂打浆,抽滤,收集滤饼,干燥,得到纯净白色固体产品。
[0039]
实施例二将nah(0.9 mmol, 3.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下
搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),继续在室温下搅拌,tlc监测反应完成。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到邻碘苯硫醚产品10,分离收率29%。
[0040]
将nah(1.5 mmol, 5.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),继续在室温下搅拌,tlc监测反应完成。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到邻碘苯硫醚产品10,分离收率73%。
[0041]
将nah(1.2 mmol, 4.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.45 mmol, 1.5 equiv,溶于0.2 ml thf),继续在室温下搅拌,tlc监测反应完成。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到邻碘苯硫醚产品10,分离收率72%。
[0042]
将nah(1.2 mmol, 4.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),继续在室温下搅拌,tlc监测反应完成。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到邻碘苯硫醚产品10,收率见上文。进行单因素变换,将所有溶剂更换为dma,得到邻碘苯硫醚产品10,分离收率54%;将所有溶剂更换为thf,得到邻碘苯硫醚产品10,分离收率69%;将所有溶剂更换为1,4
‑
二氧六环,得不到产物10;将所有溶剂更换为dme,得到邻碘苯硫醚产品10,分离收率51%;将thf(0.8 ml)更换为thf(0.4 ml),即thf:dma = 3:1,得到邻碘苯硫醚产品10,分离收率72%;将thf更换为dme,即dme:dma = 5:1,得到邻碘苯硫醚产品10,分离收率57%。
[0043]
将nah(1.2 mmol, 4.0 equiv)更换为同当量的kh、lih或者cah2称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),继续在室温下搅拌24小时,然后加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,都得不到邻碘苯硫醚产品10。用buli (2.5 equiv)、
‑
78℃替换nah(4.0 equiv)、室温,得到邻碘苯硫醚产品10,分离收率36%。
[0044]
将nah(1.2 mmol, 4.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),在0℃下搅拌12小时,得不到邻碘苯硫醚产品10。
[0045]
将nah(1.2 mmol, 4.0 equiv)称量于反应瓶中,悬于无水thf(0.8 ml thf)中搅
拌,在搅拌过程中滴加硫脲9a(0.3 mmol, 1.0 equiv,溶于0.2 ml dma),加完后在室温下搅拌1min,然后加入二碘苯2a(0.6 mmol, 2.0 equiv,溶于0.2 ml thf),在40℃下搅拌,tlc监测反应完成(1小时)。反应完成后,加入冰水和四氢呋喃淬灭反应,乙酸乙酯萃取3次,合并有机层,用饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,旋干溶剂,加入适量硅胶粉拌样,快速柱层析分离,得到邻碘苯硫醚产品10,分离收率58%。
[0046]
本发明利用大量结构不同的硫脲与邻二碘苯的反应,短时间内成功合成了丰富多样的s
‑
(2
‑
碘芳基)异硫脲,进一步的,公开了一些复杂药物分子的s
‑
芳基化反应,检验该方法在各类药物分子中的官能团耐受性,结果令人十分满意,多个经过修饰的复杂药物分子都能通过邻二碘苯生成s
‑
邻碘芳基化的产物。所有的产品都通过一系列的核磁共振技术进行结构表征,给出图1为示意,且10n的单晶结构通过x射线晶体学分析确认(图2)。可以确定硫脲类底物与二碘苯间的反应进行了c
‑
s偶联,且c
‑
s键苯环的邻位带有一个碘。
[0047]
本发明使用邻二碘苯,在nah作用下与硫脲化合物进行亲核加成反应,实现了首次利用苯炔直接与硫脲进行c
‑
s偶联生成s
‑
(2
‑
碘芳基)异硫脲,且邻位取代的二碘苯与硫脲反应具有很好的区域选择性。另外,本发明产物含有卤素,而且部分产物带有可反应基团,比如烯键,可作为工程材料的阻燃改性剂。该方案操作十分简便,无需金属催化,原料廉价易得,官能团耐受性好,为s
‑
芳基异硫脲砌块的药物合成提供了一种优秀方案,对未来的药物合成以及功能小分子发展具有重大意义。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。