1.本发明涉及有机合成技术领域,尤其涉及吡咯并酮稠环衍生物及其合成方法与应用。
背景技术:
2.酮或酚酮是一类具有羰基的非苯类7元芳香单元,是多种天然产物如秋水仙碱、茶黄素、帕瑞托酮的核心骨架,具有多种生物活性,如抗有丝分裂、抗病原体、抗癌、抗病毒和抗真菌活性。因此,从1950年代到1960年代,人们对酮或酚酮的合成进行了广泛的研究。早期的合成策略侧重于通过用各种化学计量的氧化剂如br2、nbs、kmno4和pcl5直接氧化将7元碳环转化为简单结构的酮或酚酮,然后构建含酮或酚酮的天然产物。事实上7元碳环,尤其是环庚三烯并不容易获得,这就限制了酮或酚酮化合物的合成。目前合成酮或酚酮最常用方法是基于苯环与卡宾的环丙烷化,形成一种强应变的环丙烷化苯环衍生物,它可以通过扩环过程产生各种7元碳环骨架,所得7元碳环为混合物,再经上述的氧化条件可以转化为酮或酚酮。从上述合成方法的总结,我们可以看到酮或酚酮的合成步骤繁琐,需要当量的氧化剂,副反应较多,难以分离。
3.此外,目前酮或酚酮结构有很多的研究已经开展,但是吡咯并酮稠环化合物这类全新化合物以及其生物活性,目前还未有相关报道。
技术实现要素:
4.有鉴于此,本发明的目的是提供吡咯并酮稠环衍生物及其制备方法与应用,使用简单易得的对亚甲基醌类化合物和对甲苯磺酰基甲基异腈作为起始原料,通过有机碱促进的多米诺过程一步操作快速合成出吡咯并酮稠环衍生物,整个合成方法不需要过渡金属或化学计量氧化剂参与,操作工艺简单;合成的吡咯并酮稠环衍生物具有抗肿瘤活性,可用作制备抗肿瘤药物。
5.本发明通过以下技术手段解决上述技术问题:
6.本发明一方面在于提供了一种吡咯并酮稠环衍生物,所述衍生物的结构通式如结构式(ⅰ):
[0007][0008]
其中,r1为叔丁基,ar为芳基或杂芳基;
[0009]
或者所述衍生物的结构式如化合物11:
[0010][0011]
作为优选的,所述ar为苯基、4
‑
甲氧基苯基、4
‑
(二甲氨基)苯基、3,4,5
‑
三甲氧基苯基、2
‑
溴苯基、1,1'
‑
联苯基、4
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧苯甲醛
‑2‑
基)苯基、4
‑
溴苯并[d][1,3]二氧醇
‑5‑
基、5
‑
甲基呋喃
‑2‑
基、噻吩
‑3‑
基。
[0012]
作为优选的,所述衍生物选自下列化合物:
[0013]
5,7
‑
二叔丁基
‑3‑
苯基环庚[b]吡咯
‑
6(1h)
‑
酮;
[0014]
5,7
‑
二叔丁基
‑3‑
(4
‑
甲氧基苯基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0015]
5,7
‑
二叔丁基
‑3‑
(4
‑
(二甲氨基)苯基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0016]
5,7
‑
二叔丁基
‑3‑
(3,4,5
‑
三甲氧基苯基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0017]3‑
(2
‑
溴苯基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮;
[0018]3‑
([1,1'
‑
联苯]
‑4‑
基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮;
[0019]
5,7
‑
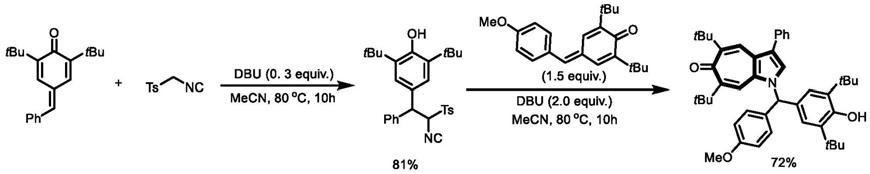
二叔丁基
‑3‑
(4
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧苯甲醛
‑2‑
基)苯基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0020]3‑
(4
‑
溴苯并[d][1,3]二氧醇
‑5‑
基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮;
[0021]
5,7
‑
二叔丁基
‑3‑
(5
‑
甲基呋喃
‑2‑
基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0022]
5,7
‑
二叔丁基
‑3‑
(噻吩
‑3‑
基)环庚[b]吡咯
‑
6(1h)
‑
酮;
[0023]
5,7
‑
二叔丁基
‑1‑
((3,5
‑
二叔丁基
‑4‑
羟基苯基)(4
‑
甲氧基苯基)甲基)
‑3‑
苯基环庚烷[b]吡咯
‑
6(1h)
‑
酮。
[0024]
本发明的另一方面在于提供了上述吡咯并酮稠环衍生物的合成方法,以所述结构式(ⅰ)为通式的化合物的合成路线如下:
[0025][0026]
所述化合物11的合成路线如下:
[0027][0028]
作为优选的,以所述结构式(ⅰ)为通式的化合物的合成方法如下:
[0029]
在干燥的反应管中,将对亚甲基醌类化合物、对甲苯磺酰基甲基异腈、1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯溶解在乙腈中,得到的反应混合物置于80℃油浴中,搅拌10小时,
通过薄层色谱监测后,将反应混合物减压浓缩,然后在硅胶柱上使用石油醚/乙酸乙酯=10/1~5/1作为洗脱剂进行分离,得到吡咯并酮稠环衍生物。
[0030]
作为优选的,所述对亚甲基醌类化合物、对甲苯磺酰基甲基异腈、1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯的摩尔比为1:2:2。
[0031]
作为优选的,所述化合物11的合成方法如下:
[0032]
s1.在干燥的反应管中,将0.2mmol的4
‑
苄叉
‑
2,6
‑
二叔丁基环己
‑
2,5
‑
二烯
‑1‑
酮、0.4mmol的1
‑
((异氰甲基)磺酰基)
‑4‑
甲苯和0.06mmol的1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯溶解在1ml乙腈中,得到的混合物置于80℃油浴中搅拌反应10h,并通过tlc监测,得到的反应物减压浓缩,在硅胶柱上使用石油醚/乙酸乙酯=10/1~5/1作为洗脱液进行分离,得到中间产物;
[0033]
s2.在干燥的反应管中,将0.16mmol的s2得到的中间产物、0.24mmol 2,6
‑
二叔丁基
‑4‑
(4
‑
甲氧基苄叉)环己
‑
2,5
‑
二烯
‑1‑
酮、0.32mmol的1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯,溶解在1ml乙腈中,随后置于80℃油浴中搅拌10小时,并通过tlc进行监测,反应完成后,真空浓缩以除去乙腈,并通过柱层析使用乙酸乙酯/己烷=5%~10%作为洗脱剂进行纯化,得到化合物11。
[0034]
另外,本发明还提供了上述吡咯并酮稠环衍生物在制备抗肿瘤药物中的应用。
[0035]
上述吡咯并酮稠环衍生物可用作制备治疗胰腺癌细胞、结肠癌细胞、前列腺癌细胞、乳腺癌细胞、头颈部鳞状细胞癌细胞的抗肿瘤药物,所述抗肿瘤药物包含吡咯并酮稠环衍生物或其药学上可接受的盐,水合物或其组合及辅料。
[0036]
本发明通过简单易得的原料,例如对甲烯醌和对甲苯磺酰甲基异氰,经有机碱促进的多米诺过程一步合成出吡咯并酮稠环衍生物,其中异氰基作为c1结构单元插入到苯酚的c
‑
c键,得到的强张力的三环降卡二烯互变异构体是不稳定的,在温和的热条件下,它会经历氮杂环亚丙基环己烯酮到酮重排,并去除对甲苯磺酰基以提供吡咯并酮衍生物。本发明的合成方法允许在单个反应中以无金属的方式形成三个c
‑
c键,从而高效地快速组装多种多样的吡咯并酮稠衍生物。该方法具有独创性涉及一系列的新的化学现象如:异腈对芳环的碳碳键插入以及氮杂环亚丙基环己烯酮到酮重排;此外我们对这类化合物进行了体外肿瘤细胞抑制活性的测试,结果显示这类化合物对人类前列腺癌、结肠癌、胰腺癌、头颈癌以及乳腺癌具有显著的抑制作用,具有抗肿瘤活性,可用于制备抗肿瘤药物。
[0037]
因此,本发明具有以下技术优势:
[0038]
(1)本发明的合成方法使用简单易得的对亚甲基醌类化合物(p
‑
qms)和对甲苯磺酰基甲基异腈(tosmic)作为起始原料,通过有机碱促进一步操作快速合成出吡咯并酮稠环衍生物,该方法所涉及的异腈对芳环的碳碳键插入以及氮杂环亚丙基环己烯酮到酮重排是全新的化学新现象,目前还未有相关报道。
[0039]
(2)本发明的吡咯并酮稠环化合物对人类前列腺癌、结肠癌、胰腺癌、头颈癌以及乳腺癌具有显著的抑制作用,尤其以化合物9的效果最显著,10μm的化合物9生长抑制率可达到90%,且试验证明化合物9对前列腺癌细胞增殖具有显著的抑制作用,本发明的吡咯并酮稠环化合物具有抗肿瘤活性,可用于制备抗肿瘤药物。
[0040]
(3)本发明的合成方法不需要过渡金属或化学计量氧化剂参与,操作工艺简单,通
过串联步骤一步即可得到吡咯并酮稠衍生物,原料简单易得。
附图说明
[0041]
图1是10μm的化合物1
‑
11对胰腺癌细胞系、结肠癌细胞系,前列腺癌细胞系、乳腺癌细胞系、头颈部鳞状细胞癌细胞系的生长能力影响图;
[0042]
图2是不同浓度的化合物9对前列腺癌细胞存活能力的影响图;
[0043]
图3是不同时间2.5μm的化合物9对前列腺癌细胞生长影响图;
[0044]
图4是化合物9对pc3细胞的克隆形成能力影响图;
[0045]
图5是化合物1的核磁图;
[0046]
图6是化合物2的核磁图;
[0047]
图7是化合物3的核磁图;
[0048]
图8是化合物4的核磁图;
[0049]
图9是化合物5的核磁图;
[0050]
图10是化合物6的核磁图;
[0051]
图11是化合物7的核磁图;
[0052]
图12是化合物8的核磁图;
[0053]
图13是化合物9的核磁图;
[0054]
图14是化合物10的核磁图;
[0055]
图15是化合物11的核磁图。
具体实施方式
[0056]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0057]
本发明的吡咯并酮稠环衍生物主要有两类,一类是以结构式(ⅰ)为结构通式的化合物:
[0058][0059]
其中,r1为叔丁基,ar为芳基或杂芳基,具体的,ar为苯基、4
‑
甲氧基苯基、4
‑
(二甲氨基)苯基、3,4,5
‑
三甲氧基苯基、2
‑
溴苯基、1,1'
‑
联苯基、4
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧苯甲醛
‑2‑
基)苯基、4
‑
溴苯并[d][1,3]二氧醇
‑5‑
基、5
‑
甲基呋喃
‑2‑
基、噻吩
‑3‑
基;
[0060]
本发明的吡咯并酮稠环衍生物的另一类的结构式如化合物11:
[0061][0062]
以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成路线如下:
[0063][0064]
具体的,以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法如下:
[0065]
在烘箱干燥的10ml反应管中,将0.2mmol的对亚甲基醌类化合物(p
‑
qms)、0.4mmol对甲苯磺酰基甲基异腈(tosmic)、60μl 0.4mmol的1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯(dbu)溶解在1ml的乙腈(mecn)中,然后将得到的反应混合物置于80℃油浴中搅拌10小时,并通过薄层色谱(tlc)监测,将反应混合物在减压下浓缩,接着在硅胶柱上使用石油醚/乙酸乙酯=10/1~5/1分离,优选使用乙酸乙酯/己烷=6/1作为洗脱剂进行分离,得到目标化合物吡咯并酮稠环衍生物。
[0066]
以下实施例中的产品检测条件如下:
[0067]
在400mhz固体核磁共振谱仪(bruker avance iii 400mhz)上,以四甲基硅(tms)作为内标记录1h和
13
c nmr。1h nmr数据报告如下:化学位移,以ppm(δ)为单位,多重性(s=单峰,d=双重峰,t=三重峰,m=多重峰),偶合常数(hz),相对强度;
13
c nmr数据报告如下:化学位移(ppm)。
[0068]
hplc
‑
ms分析在shimadzu
‑
2020lc
‑
ms仪器上使用以下条件进行:shim
‑
pack vpods c18色谱柱(反相,150
×
2.0mm);在10.0分钟内使用80%乙腈和20%水;流速为0.4ml/min;紫外光电二极管阵列检测范围为200至300nm。产物通过biotage isolera
tm spektra系统和己烷/乙酸乙酯溶剂系统纯化。所有试剂和溶剂均购自商业渠道,无需进一步纯化即可使用。
[0069]
为了更清楚的了解本发明的吡咯并酮稠环衍生物,进行了以下11个实施例的试验。
[0070]
实施例1
[0071]
本实施例的吡咯并酮稠环衍生物为化合物1:5,7
‑
二叔丁基
‑3‑
苯基环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物1的化学结构式如下:
[0072][0073]
本实施例的化合物1的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的
合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄色固体化合物1,熔点219
‑
223℃,产率为88%。化合物1的核磁图如图5所示。
[0074]1h nmr(400mhz,cdcl3)δ8.80(s,1h),7.62(s,1h),7.45(d,j=4.3hz,4h),7.35
–
7.30(m,2h),7.06(d,j=2.5hz,1h),1.42(s,9h),1.39(s,9h)ppm;
13
c nmr(100mhz,cdcl3)δ195.4,148.2,147.1,134.6,132.0,128.8,128.6 126.6,125.2,123.5,121.0,119.2,118.8,38.2,38.1,31.9,31.7ppm;hrms(esi)m/z calcd for c
23
h
28
no
(m h)
334.2165,found m/z 334.2160.
[0075]
实施例2
[0076]
本实施例的吡咯并酮稠环衍生物为化合物2:5,7
‑
二叔丁基
‑3‑
(4
‑
甲氧基苯基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物2的化学结构式如下:
[0077][0078]
本实施例的化合物2的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为4
‑
甲氧基苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到淡黄色固体化合物2,熔点202
‑
205℃,产率为86%。化合物2的核磁图如图6所示。
[0079]1h nmr(400mhz,cdcl3)δ8.77(s,1h),7.59(s,1h),7.36(d,j=8.6hz,2h),7.31(s,1h),7.00(d,j=8.4hz,3h),3.86(s,3h),1.42(s,9h),1.39(s,9h)ppm;
13
c nmr(100mhz,cdcl3)δ195.3,158.5,148.1,146.9,131.9,129.7,127.0,124.9,123.6,121.1,119.2,118.4,114.2,55.4,38.1,38.1,31.9,31.7ppm;hrms(esi)m/z calcd for c
24
h
30
no
2
(m h)
364.2271,found m/z 364.2282.
[0080]
实施例3
[0081]
本实施例的吡咯并酮稠环衍生物为化合物3:5,7
‑
二叔丁基
‑3‑
(4
‑
(二甲氨基)苯基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物3的化学结构式如下:
[0082][0083]
本实施例的化合物3的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为4
‑
(二甲氨基)苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到棕色固体化合物3,熔点147
‑
150℃,产率为79%。化合物3的核磁图如图7所示。
[0084]1h nmr(400mhz,cdcl3)δ8.58(s,1h),7.65(s,1h),7.34(d,j=8.5hz,2h),7.30(s,
1h),7.00(d,j=2.4hz,1h),6.84(d,j=8.5hz,2h),3.00(s,6h),1.42(s,9h),1.39(s,9h)ppm;
13
cnmr(100mhz,cdcl3)δ195.2,149.5,147.9,146.7,131.8,129.4,125.5,123.9,122.7,121.1,119.2,118.0,112.9,40.7,38.1,38.1,31.9,31.7ppm;hrms(esi)m/z calcd for c
25
h
33
n2o
(m h)
377.2587,found m/z 377.2584.
[0085]
实施例4
[0086]
本实施例的吡咯并酮稠环衍生物为化合物4:5,7
‑
二叔丁基
‑3‑
(3,4,5
‑
三甲氧基苯基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物4的化学结构式如下:
[0087][0088]
本实施例的化合物4的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为3,4,5
‑
三甲氧基苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄棕色固体化合物4,熔点205
‑
208℃,产率为80%。化合物4的核磁图如图8所示。
[0089]1h nmr(400mhz,cdcl3)δ9.04(s,1h),7.67(s,1h),7.34(s,1h),7.08(d,j=2.5hz,1h),6.67(s,2h),3.92(s,3h),3.90(s,6h),1.41(d,j=5.7hz,18h)ppm;
13
c nmr(100mhz,cdcl3)δ195.4,153.4,148.4,147.0,136.8,132.1,130.4,125.1,123.5,120.8,119.2,118.6,105.6,61.0,56.1,38.1,38.1,32.0,31.7ppm;hrms(esi)m/z calcd for c
26
h
34
no
4
(m h)
424.2482,found m/z 424.2477.
[0090]
实施例5
[0091]
本实施例的吡咯并酮稠环衍生物为化合物5:3
‑
(2
‑
溴苯基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物5的化学结构式如下:
[0092][0093]
本实施例的化合物5的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为2
‑
溴苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄棕色固体化合物5,熔点210
‑
215℃,产率为77%。化合物5的核磁图如图9所示。
[0094]1h nmr(400mhz,dmso)δ11.75(s,1h),7.70(d,j=7.9hz,1h),7.45(s,1h),7.39(t,j=7.3hz,1h),7.31(d,j=7.4hz,1h),7.27
–
7.20(m,2h),7.04(s,1h),1.31(s,9h),1.19(s,9h)ppm;
13
c nmr(100mhz,dmso)δ194.2,146.7,145.5,135.6,133.4,133.2,131.8,129.5,128.2,124.2,124.2,122.6,122.0,121.2,120.2,38.1,38.0,31.8,31.8ppm;hrms(esi)m/z calcd for c
23
h
27
brno
(m h)
412.1271,found m/z 412.1284.
[0095]
实施例6
[0096]
本实施例的吡咯并酮稠环衍生物为化合物6:3
‑
([1,1'
‑
联苯]
‑4‑
基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物6的化学结构式如下:
[0097][0098]
本实施例的化合物6的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为1,1'
‑
联苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄色固体化合物6,熔点163
‑
166℃,产率为73%。化合物6的核磁图如图10所示。
[0099]1h nmr(400mhz,cdcl3)δ8.64(s,1h),7.68(dd,j=10.7,7.1hz,5h),7.53(d,j=8.1hz,2h),7.48(dd,j=12.8,5.2hz,2h),7.37(d,j=7.3hz,1h),7.32(s,1h),7.12(d,j=2.4hz,1h),1.44(s,9h),1.41(s,9h)ppm;
13
c nmr(100mhz,cdcl3)δ195.3,148.4,147.2,140.8,139.4,133.6,132.1,128.9,128.8,127.5,127.3,127.0,124.9,123.4,121.0,119.0,118.7,38.2,38.1,31.9,31.7ppm;hrms(esi)m/z calcd for c
29
h
32
no
(m h)
410.2478,found m/z 410.2473.
[0100]
实施例7
[0101]
本实施例的吡咯并酮稠环衍生物为化合物7:5,7
‑
二叔丁基
‑3‑
(4
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧苯甲醛
‑2‑
基)苯基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物7的化学结构式如下:
[0102][0103]
本实施例的化合物7的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为4
‑
(4,4,5,5
‑
四甲基
‑
1,3,2
‑
二氧苯甲醛
‑2‑
基)苯基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄色固体化合物7,熔点224
‑
226℃,产率为59%。化合物7的核磁图如图11所示。
[0104]1h nmr(400mhz,cdcl3)δ8.63(s,1h),7.89(d,j=7.7hz,2h),7.62(s,1h),7.47(d,j=7.7hz,2h),7.29(s,1h),7.11(d,j=2.4hz,1h),1.42(s,9h),1.37(s,21h)ppm;
13
c nmr(100mhz,cdcl3)δ195.2,148.5,147.3,137.4,135.2,132.1,127.8,125.2,123.4,118.9,118.8,83.8,38.1,38.1,31.9,31.7,24.9ppm;hrms(esi)m/z calcd for c
29
h
39
bno
3
(m h)
460.3018,found m/z 460.3024.
[0105]
实施例8
[0106]
本实施例的吡咯并酮稠环衍生物为化合物8:3
‑
(4
‑
溴苯并[d][1,3]二氧醇
‑5‑
基)
‑
5,7
‑
二叔丁基环庚烷[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物8的化学结构式如下:
[0107][0108]
本实施例的化合物8的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为4
‑
溴苯并[d][1,3]二氧醇
‑5‑
基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到黄色固体化合物8,熔点212
‑
216℃,产率为96%。化合物8的核磁图如图12所示。
[0109]1h nmr(400mhz,dmso)δ11.73(s,1h),7.54
–
7.47(m,1h),7.38
–
7.31(m,1h),7.22(dd,j=4.1,2.7hz,1h),7.17
–
7.07(m,1h),6.97
–
6.87(m,1h),6.18
–
6.09(m,2h),1.37(s,9h),1.27(s,9h)ppm;
13
c nmr(100mhz,dmso)δ194.1,148.1,147.6,146.6,145.4,131.6,128.6,124.3,122.8,121.9,121.3,120.2,114.8,113.0,112.4,102.6,38.1,37.9,31.9,31.8ppm;hrms(esi)m/z calcd for c
24
h
27
brno
3
(m h)
456.1169,found m/z 456.1167.
[0110]
实施例9
[0111]
本实施例的吡咯并酮稠环衍生物为化合物9:5,7
‑
二叔丁基
‑3‑
(5
‑
甲基呋喃
‑2‑
基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物9的化学结构式如下:
[0112][0113]
本实施例的化合物9的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为5
‑
甲基呋喃
‑2‑
基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到棕黄色固体化合物9,熔点130
‑
134℃,产率为72%。化合物9的核磁图如图13所示。
[0114]1h nmr(400mhz,cdcl3)δ8.65(s,1h),7.83(s,1h),7.26(s,1h),7.21(d,j=2.6hz,1h),6.30(d,j=3.0hz,1h),6.08(d,j=2.8hz,1h),2.37(s,3h),1.44(s,9h),1.41(s,9h)ppm;
13
cnmr(100mhz,cdcl3)δ195.1,150.6,148.5,147.7,147.3,131.8,123.6,119.8,118.8,117.8,115.5,107.1,105.9,38.2,38.1,31.9,31.6,13.6ppm;hrms(esi)m/z calcd for c
22
h
28
no
2
(m h)
338.2115,found m/z 338.2112.
[0115]
实施例10
[0116]
本实施例的吡咯并酮稠环衍生物为化合物10:5,7
‑
二叔丁基
‑3‑
(噻吩
‑3‑
基)环庚[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物10的化学结构式如下:
[0117][0118]
本实施例的化合物10的合成按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,其中,对亚甲基醌类化合物中的r1为叔丁基,ar为5
‑
甲基呋喃
‑2‑
基。具体合成方法、原料配方及工艺条件按照以结构式(ⅰ)为通式的吡咯并酮稠环衍生物的合成方法进行,最后得到棕黄色固体化合物10,熔点104
‑
110℃,产率为64%。化合物10的核磁图如图14所示。
[0119]1h nmr(400mhz,cdcl3)δ8.78(s,1h),7.78(s,1h),7.29(d,j=3.6hz,2h),7.13(dd,j=5.5,2.9hz,2h),7.11
–
7.07(m,1h),1.42(d,j=2.8hz,18h)ppm;
13
c nmr(100mhz,cdcl3)δ195.2,148.6,147.4,136.4,132.1,127.7,124.4,124.0,123.3,120.9,119.1,118.9,118.0,38.2,38.1,31.9,31.7ppm;hrms(esi)m/z calcd for c
21
h
26
nos
(m h)
340.1730,found m/z 340.1728.
[0120]
实施例11
[0121]
本实施例的吡咯并酮稠环衍生物为化合物11:5,7
‑
二叔丁基
‑1‑
((3,5
‑
二叔丁基
‑4‑
羟基苯基)(4
‑
甲氧基苯基)甲基)
‑3‑
苯基环庚烷[b]吡咯
‑
6(1h)
‑
酮,具体的,化合物11的化学结构式如下:
[0122][0123]
本实施例的化合物11的合成路线如下:
[0124][0125]
具体的,化合物11的合成方法如下:
[0126]
s1:在烘箱干燥的10ml反应管中,将0.2mmol的4
‑
苄叉
‑
2,6
‑
二叔丁基环己
‑
2,5
‑
二烯
‑1‑
酮、0.4mmol的1
‑
((异氰甲基)磺酰基)
‑4‑
甲苯、0.06mmol的1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯(dbu)溶解在1ml乙腈(mecn)中,将混合物置于80℃油浴中搅拌反应10h,并通过tlc监测。然后在减压下浓缩反应混合物,在硅胶柱上使用石油醚/乙酸乙酯=10/1~5/1作为洗脱液进行分离,得79mg中间产物,产率81%。
[0127]
s2:在烘箱干燥的10ml反应管中,将79mg 0.16mmol的中间产物、78mg 0.24mmol的2,6
‑
二叔丁基
‑4‑
(4
‑
甲氧基苄叉)环己
‑
2,5
‑
二烯
‑1‑
酮、49ul 0.32mmol的1,8
‑
二氮杂双环[5.4.0]十一碳
‑7‑
烯(dbu)溶解在1ml乙腈(mecn)中,将混合物置于80℃油浴中搅拌10小
时,并通过tlc进行监测。反应完成后,在真空下浓缩混合物以除去乙腈,并通过柱层析(乙酸乙酯/己烷=5%~10%,本实施例使用乙酸乙酯/己烷=6%)纯化残余物,得到目标76mg黄色固体化合物11,其熔点是223
‑
226℃产率为72%。化合物11的核磁图如图15所示。
[0128]1h nmr(400mhz,cdcl3)δ7.63(s,1h),7.40(q,j=7.4hz,4h),7.32(s,1h),7.30(d,j=7.3hz,1h),7.07(d,j=8.4hz,2h),6.95(s,2h),6.89(d,j=8.4hz,2h),6.65(s,1h),6.61(s,1h),5.25(s,1h),3.81(s,3h),1.38(s,9h),1.36(s,18h),1.27(s,9h)ppm;
13
c nmr(100mhz,cdcl3)δ194.9,159.2,153.6,147.1,147.0,136.3,134.9,132.2,131.6,129.6,129.5,128.6,126.3,125.2,123.8,122.8,122.4,122.3,118.2,114.1,64.7,55.3,38.3,38.1,34.4,31.8,31.6,30.2ppm;hrms(esi)m/z calcd for c
45
h
56
no
3
(m h)
658.4255,found m/z 658.4254.
[0129]
实施例12
[0130]
本实施例对化合物1
‑
11进行了抗肿瘤活性检测,选用人源肿瘤细胞系aspc
‑
1,hct116,pc3,mda
‑
mb
‑
231,scc
‑
15,cal33,这些细胞系均购自美国特种培养物收集中心(american type culture collection,atcc)。aspc
‑
1细胞在rpmi
‑
1640培养基中培养,hct116在mccoy's5a培养基中培养,pc3在f12k培养基中培养,mda
‑
mb
‑
231、scc
‑
15、cal33在dmem培养基中培养。所有培养基补充10%胎牛血清(fbs)以提供细胞生长所需生长因子,细胞培养条件为37℃、含5%co2的恒温培养箱。培养步骤如下:
[0131]
(1)将处于对数生长期的肿瘤细胞用胰酶消化后,用完全培养基重悬,用细胞计数仪测定细胞浓度后,将细胞用对应的完全培养基稀释到3
×
104个/ml;
[0132]
(2)在96孔板的每个孔中接种100μl细胞悬液,置于细胞培养箱中过夜孵育;
[0133]
(3)将化合物1
‑
11用对应细胞的完全培养基稀释至20μm,取100μl化合物稀释液分别加入96孔板的每个孔中,使化合物终浓度为10μm,每个化合物加3个复孔,对照组为与化合物等体积的dmso,将细胞放回细胞培养箱中培养72小时;
[0134]
(4)通过mtt实验检测化合物1
‑
11对各肿瘤细胞存活能力的抑制作用,分布用化合物1
‑
11各自处理肿瘤细胞72小时后,每个孔中加入20μl浓度为5mg/ml的mtt溶液,37℃继续孵育4小时;
[0135]
(5)弃去细胞培养液,加入150μl dmso将细胞溶解,用酶标仪检测570nm出的od值(od570);
[0136]
(6)处理数据,根据化合物处理组与对照处理组的od570计算相对存活率,细胞存活率=od570
化合物
/od570
dmso
。
[0137]
化合物1
‑
11对各肿瘤细胞的抑制率如表1所示。
[0138]
表1吡咯并酮稠环衍生物的抗肿瘤活性
[0139][0140]
实验结果:10μm的化合物1
‑
11对胰腺癌细胞系、结肠癌细胞系,前列腺癌细胞系、乳腺癌细胞系、头颈部鳞状细胞癌细胞系的生长能力影响如图1所示。图1数据表明,化合物1
‑
11对胰腺癌细胞、结肠癌细胞、前列腺癌细胞、乳腺癌细胞、头颈部鳞状细胞癌细胞的生长能力均有一定的抑制作用,尤其是化合物9对所测试的6种肿瘤细胞生长能力均表现出良好的抑制效果。实验结果表明,本发明描述的化合物9可抑制肿瘤细胞生长,具有良好的抗肿瘤活性,可应用于制备抗肿瘤药物,尤其是针对前列腺癌的抗肿瘤药物。
[0141]
为了检测不同浓度的化合物9对前列腺癌细胞存活能力的影响,将pc3、du145细胞分别用含10%fbs的f12k培养基、dmem培养基进行稀释,使细胞终浓度为2
×
104个/ml,然后分别接种100μl细胞悬液到96孔板中,待细胞贴壁后加入化合物9使其终浓度为0μm、1.25μm、2.5μm、5μm、10μm、20μm,每个浓度加5个复孔,化合物处理72小时后,用mtt法检测不同浓度化合物9对pc3、du145细胞存活能力的影响。实验结果如图2所示,随着化合物浓度升高,两株前列腺癌细胞存活能力明显降低,表明化合物9对两种前列腺癌肿瘤细胞均具有一定的抑制活性,且在pc3细胞中活性较好。化合物9对前列腺癌细胞pc3和du145的半数致死浓度分别为2.1μm和6.2μm。
[0142]
为了更加直观真实的反应化合物9对前列腺癌细胞生长的抑制作用,通过高内涵分析系统对培养肿瘤细胞进行实时拍照,从而检测化合物9对前列腺癌细胞生长的抑制作用。如图3所示,2.5μm化合物9分别处理pc3和du145细胞后,随着时间的延续,pc3细胞数量变化不明显,du145细胞细胞数量微弱上升,细胞增殖受到抑制。而对照组细胞数量显著上升,细胞增殖正常。表明2.5μm化合物9能够明显抑制前列腺癌细胞pc3和du45的增殖。
[0143]
为了进一步验证化合物9对前列腺癌细胞增殖能力的抑制作用,在图3实验的基础上,进行克隆形成实验。将1
×
103个pc3细胞接种到6孔板里,在37℃,5%的二氧化碳细胞培养箱中培养过夜。分别加入0μm、1.25μm与2.5μm的化合物9处理细胞48小时,然后换新鲜的完全培养基继续培养4天。用4%的多聚甲醛对细胞进行固定并用1%的结晶紫溶液染色,检测化合物9对前列腺癌细胞克隆形成能力的影响。结果如图4所示,化合物9可以显著抑制pc3细胞的克隆形成能力。进一步表明化合物9可抑制前列腺癌细胞增殖。
[0144]
通过上述实验,证明化合物9对前列腺癌细胞增殖具有显著的抑制作用。
[0145]
因此,本发明的吡咯并酮稠环衍生物可用于制备抗肿瘤药物,具体的,可用作制备治疗胰腺癌细胞、结肠癌细胞、前列腺癌细胞、乳腺癌细胞、头颈部鳞状细胞癌细胞的抗肿瘤药物,上述抗肿瘤药物包含吡咯并酮稠环衍生物或其药学上可接受的盐,水合物或其组合及辅料。
[0146]
以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。本发明未详细描述的技术、形状、构造部分均为公知技术。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。