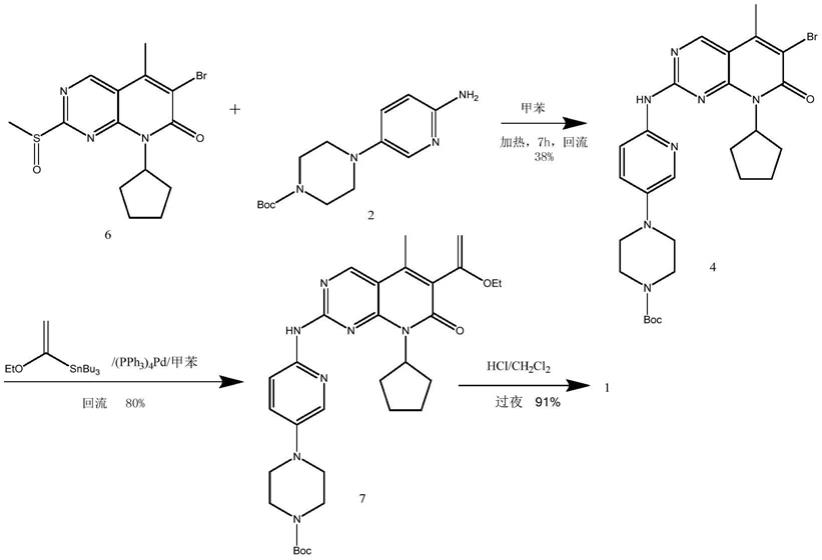
1.本发明属于化学合成技术领域,具体涉及一种帕布昔利布的低成本制备方法。
背景技术:
2.帕布昔利布(palbociclib)是由美国辉瑞研发的治疗乳腺癌新药,商品名称为ibrance,是一种口服细胞周期素依赖性激酶(cdks)4和6抑制剂,为美国fda批准首个细胞周期蛋白依赖性激酶4/6(cdk4/6)抑制剂。cdks4和6是细胞周期的关键调节因素,其能够触发细胞周期进展。ibrance在美国的适应症为联合来曲唑用于治疗雌激素受体阳性,人类表皮生长因子受体2阴性(er /her2
‑
)绝经后晚期乳腺癌患者,作为初始的内分泌治疗为基础的方案治疗转移性疾病。
3.帕布昔利布,中文名:6
‑
乙酰基
‑8‑
环戊基
‑5‑
甲基
‑2‑
[[5
‑
(哌嗪
‑1‑
基)吡啶
‑2‑
基]氨基]
‑
8h
‑
吡啶并[2,3
‑
d]嘧啶
‑7‑
酮,结构式如下所示。
[0004][0005]
文献报道的帕布昔利布的合成方法主要有以下几种:
[0006]
方法一:mark barvian等人在美国专利us6936612b2中以6
‑
溴
‑8‑
环戊基
‑2‑
甲基亚磺酰基
‑5‑
甲基
‑
8h
‑
吡啶并[2,3
‑
d]嘧啶
‑7‑
酮(式6)为原料,在甲苯中与4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式2)通过n
‑
烷基化反应,得到化合物4,化合物4经(三苯基膦)钯催化与三丁基(1
‑
乙氧基乙烯)锡进行stille偶联反应合成化合物7,再在二氯甲烷体系中进行水解和脱保护得到目标产物帕布昔利布,具体的合成路线如下。文献还报道了以4
‑
氯
‑2‑
甲硫基嘧啶
‑5‑
甲酸乙酯为起始原料经5步反应制备化合物6的方法。
[0007][0008]
此路线制备化合物4的收率偏低,仅为38%,制备化合物7的stille偶联反应,用到有机锡等剧毒物质,不利于工业化生产。
[0009]
方法二:brian等人在专利wo2008032157a2中报道了以4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式2)和6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(式3)为起始原料,以六甲基二硅基氨基锂(lihmds)为碱进行n
‑
烷基化得到化合物4,化合物4与正丁基乙烯基醚在[双(二苯基膦基二茂铁)]二氯化钯(pd(dppf)2cl2)的催化下进行herk偶联反应得到化合物5,最后经羟基乙磺酸催化进行重排和水解反应得到目标产物帕布昔利布,反应路线如下:
[0010][0011]
与路线1相比,其关键中间体3在嘧啶环的2位以氯原子取代了亚甲磺酰基,增加了嘧啶环2位的活性,其次,关键中间体3与中间体2经n
‑
烷基化反应获得化合物4时用lihmds为碱,提高反应收率至92%。另外,文献采用廉价易得的2,4
‑
二氯
‑5‑
溴嘧啶,经3步反应得到化合物3,大大降低了原材料成本和可工业化能力。
[0012]
但路线2采用[双(二苯基膦基二茂铁)]二氯化钯(pd(dppf)2cl2),其用量约为化合物4的3%,随着钯价格的日益上涨,造成帕布昔利布成本的不断上涨,按照文献工艺核算成本,钯的价格已经成为该工艺的主要成本,远超原材料成本。
[0013]
申请人在中国专利cn112898299a和中国专利cn112661753a中,对化合物3的合成进行了优化和改进,以实现工业化、低成本生产。
[0014]
erdman等人在专利wo 2014128588a1中采用醋酸钯做催化剂,双(2
‑
二苯基磷苯基)醚(dpephos)为配体,化合物4与正丁基乙烯基醚herk偶联反应,醋酸钯的用量为化合物4质量的1.6%,钯催化剂的价格仍然是制约帕布昔利布推广使用的关键因素。
[0015]
作为重磅炸弹级新药,帕布昔利布的用量很大。随着中国政府和医药化工企业对环境保护意识的逐年增强,开发一种低成本、高收率、环境友好的帕布昔利布工业化合成方法为当前需要解决的问题。
技术实现要素:
[0016]
本发明克服了上述现有技术的不足,提供一种帕布昔利布的低成本制备方法。该方法以4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯为起始原料,以大位阻碱为缚酸剂,与6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮发生亲核取代反应,后处理通过淬灭、脱碱得大颗粒式4化合物,式4化合物以正丁醇、水为溶剂,二异丙基乙胺为缚酸剂和保护剂,在复合催化剂氯化钯和碘化亚铜的作用下,与正丁基乙烯基醚发生herk烷基化反应,在有机碱的保护下,通过酯类溶剂精制高收率的得到高纯度的式5化合物,式5化合物通
过正丁醇、苯甲醚、水混合溶剂,酸性条件下水解,直接高收率的得到高纯度的帕布昔利布成品。该方法大大降低了钯催化剂的使用量,且操作简便、对环境污染少、收率高、成本低、产品质量高,更适合于工业化生产。
[0017]
本发明的技术方案是:一种帕布昔利布的低成本制备方法,包括:
[0018]
s1:4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式2)在甲苯中,通过有机碱作为缚酸剂,与6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(式3)发生氨基化反应,经后处理得到式4化合物;
[0019]
s2:以正丁醇为溶剂,化合物4与正丁基乙烯基醚在钯催化剂催化下进行herk烷基化反应得到化合物5;
[0020]
s3:化合物5水解反应得到目标产物帕布昔利布;
[0021]
其特征是,
[0022]
所述步骤s1的有机碱为大位阻金属有机碱。
[0023]
所述步骤s2为:以氯化钯和碘化亚铜作为催化剂,双(2
‑
二苯基磷苯基)醚和三苯基膦作为配合物,正丁醇和水为溶剂,二异丙基乙胺作碱,与正丁基乙烯基醚进行herk烷基化反应。
[0024]
进一步的,所述步骤s3为:在正丁醇、苯甲醚、水的三相体系中,式5化合物用酸水解。
[0025]
合成路线如下所示:
[0026][0027]
其中,lihmds:六甲基二硅基氨基锂,dpephos:双(2
‑
二苯基磷苯基)醚,diea:二异丙基乙胺。
[0028]
更优选的,本发明具体包括以下以下步骤:
[0029]
s1:4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式2)在甲苯中,通过大位阻金属有机碱作为缚酸剂,与6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(式3)发生氨基化反应,经灭活,脱碱处理,得到式4化合物;
[0030]
s2:氮气保护下,式4化合物以氯化钯和碘化亚铜作为催化剂,双(2
‑
二苯基磷苯基)醚和三苯基膦作为配合物,正丁醇和水为溶剂,二异丙基乙胺作碱,与正丁基乙烯基醚进行herk烷基化反应,得到式5化合物粗品,再通过乙酸乙酯或醋酸异丙酯作溶剂,有机碱
为稳定剂,通过精制(加热回流,降温析晶)得到高纯度的式5化合物;
[0031]
s3:在正丁醇、苯甲醚、水的三相体系中,式5化合物用酸水解,再通过中和、水洗脱盐、降温结晶得到高纯度(纯度99.9以上,单杂0.05%)的帕布昔利布(式1)。
[0032]
其中,
[0033]
步骤s1中的大位阻金属有机碱为:二异丙基氨基锂、六甲基二硅基氨基锂、六甲基二硅基氨基钠、六甲基二硅基氨基钾、叔丁醇钠、叔丁醇钾中的一种,优选六甲基二硅基氨基钠。该步骤主要杂质为4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯(式2)的吡啶环杂质(式8),反应路线如下:
[0034][0035]
式8所示的杂质是造成路线1收率仅为38%的主要因素,因此,采用大位阻碱,首先脱除4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯的吡啶环6位氢,增强6位氨基的亲和性,再与6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(式3)反应,不仅降低其反应活化能,更减少了杂质8的产生,提高了式4化合物的收率和纯度。
[0036]
步骤s1中的灭活方式为:加入丙酮与水体积比1.8~2.2:1的混合溶剂,得到大颗粒的式4化合物粗品。
[0037]
步骤s1中脱碱处理为:将所得固体加入到二氯甲烷与水的两相体系中,采用醋酸调ph值脱除物料中影响herk反应残留的有机碱。有机碱主要为残留的六甲基二硅氮烷,具有一定的还原性,残留量可使氯化钯失活,造成下一步herk反应消耗钯量增加。
[0038]
步骤s2中的催化剂氯化钯、碘化亚铜与式4化合物的质量比为0.0001~0.0005:0.005~0.02:1,优选质量比为0.0002:0.01:1。通过加入碘化亚铜,将氯化钯用量减为专利wo 2014128588a1实例描述用量(文献用醋酸钯量为中间体i的1.6%)的1.5%,大大减少了贵金属钯的用量,这是我们对工艺的最大改进,贵金属钯的成本,从占帕布昔利布主要成本到微不足道,极大降低了帕布昔利布的生产成本。
[0039]
步骤s2中的配合物双(2
‑
二苯基膦基苯基)醚、三苯基膦与化合物4的质量比为0.0005~0.002:0.01~0.05:1,优选质量比为0.001:0.02:1。正丁基乙烯醚、二异丙基乙胺与化合物4的摩尔比为1.2~4:1.2~2.0:1,优选1.5:1.5:1。正丁醇和水的体积比为5~15:1,优选10:1。
[0040]
步骤s2中的精制过程,稳定剂有机碱为三乙胺、二异丙基乙胺、吡啶、n
‑
甲基吗啉、4
‑
二甲氨基吡啶等有机碱中的一种,其加入量为化合物5粗品质量的0.5~10%,优选2%。原研公司在专利cn105008357a中描述该步骤收率仅为79%,经研究,造成收率低的主要原因,是其后处理过程中产生了易溶于有机溶剂的丁氧乙烯基降解杂质(式9):
[0041][0042]
丁氧乙烯基在弱酸性中即可快速降解,但在碱存在下,降解速率缓慢,通过添加有机碱的方式,通过乙酸乙酯或醋酸异丙酯精制,可得到高纯度的(≥99%)的式5化合物,降解杂质(式9)仅为0.1~0.3%。
[0043]
步骤s3中的正丁醇、苯甲醚、水和式5化合物的质量比8~12:12~18:8~12:1,优选10:15:10:1。
[0044]
步骤s3中的酸为盐酸或氢溴酸,用量为式5化合物的1.2~3当量,优选1.5当量。
[0045]
步骤s3中的中和剂为氢氧化钠溶液、氢氧化钾溶液或氨水中的一种,优选氢氧化钠溶液。
[0046]
步骤s3中的水洗脱盐为70~90℃。
[0047]
步骤s1中的反应温度为20~30℃;步骤s2中的反应温度为90~100℃;步骤s3中的反应温度为75~85℃。
[0048]
本发明的技术特点和优益效果:
[0049]
1、本发明利用4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯为起始原料,以甲苯为溶剂,以大位阻碱为缚酸剂,与6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(制备方法见中国专利cn112898299a和中国专利cn112661753a)发生亲核取代反应,反应条件温和,经丙酮/水淬灭得到易于过滤的大颗粒式4化合物,后处理简单,产品收率高,质量好,适合工业化生产。
[0050]
2、本发明采用正丁醇和水为溶剂,二异丙基乙胺为碱和保护剂,式4化合物与正丁基乙烯基醚在复合催化剂氯化钯和碘化亚铜,复合配体三苯基膦和(2
‑
二苯基磷苯基)醚存在下,进行herk反应,经乙酸乙酯或醋酸异丙酯,在有机碱做稳定剂的条件下,高收率的得到高纯度的式5化合物。该步骤的最大特点是通过添加碘化亚铜和溶剂水,将贵金属钯用量减少为文献用量的1.5%,极大降低了原材料成本和贵金属钯对环境的污染。而通过精制得到的式5化合物,纯度高,通过步骤3直接水解、中和、脱盐处理,不用再通过精制即可得到高纯度的帕布昔利布成品,简化了反应步骤,降低了原材料成本。
[0051]
3、本发明采用三相体系进行水解步骤,仅仅是为了简化工艺,可以通过反应、后处理、脱盐步骤直接得到合格的帕布昔利布,从而简化工业生产操作步骤,提高工作效率。
[0052]
综上,本发明为帕布昔利布原料药的合成,提供了一条高质量、低成本、对环境友好、适合工业化生产的制备方法,尤其是大大降低了钯催化剂的使用量。本发明的低成本制备方法有利于帕布昔利布制剂的使用和推广,同时,有利于减少患者的资金支出,使更多的乳腺癌患者能够用上安全性高的帕布昔利布靶向药。
具体实施方式
[0053]
下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。
[0054]
帕布昔利布检测的色谱条件:
[0055]
色谱柱:hypersil bds c18(4.6
×
250mm,5μm);
[0056]
uv检测器(检测波长220nm);
[0057]
流动相a:称取2.875g磷酸二氢铵到1l水中,用磷酸调ph至3.7;流动相b:乙腈;
[0058]
流速:1ml/min;
[0059]
进样量:10μl;
[0060]
运行时间:55min;
[0061]
稀释液:乙腈;
[0062]
柱温:30℃
[0063]
梯度洗脱条件如下表1。
[0064]
表1梯度洗脱程序
[0065]
时间(min)流动相a流动相b08020302080402080418020558020
[0066]
实施例1:4
‑
(6
‑
((6
‑
溴
‑8‑
环戊基
‑
7,8
‑
二氢
‑5‑
甲基
‑7‑
氧代吡啶并[2,3
‑
d]嘧啶
‑2‑
基)氨基)
‑3‑
吡啶基)
‑1‑
哌嗪羧酸叔丁酯(式4化合物)的实验室制备
[0067]
5l反应瓶中,加入167g(0.6mol)4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,1600ml甲苯,通氮气保护,于20~30℃滴加750ml 1mol/l的六甲基二硅基氨基钠四氢呋喃溶液,保持20~30℃搅拌反应30分钟。将171g(0.5mol)6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮分散在600ml甲苯中,缓慢倒入反应液,加毕,保持30℃反应4h,tlc检测6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮消失为反应终点(展开剂:乙酸乙酯/正己烷=1/4)。反应毕,缓慢加入340ml丙酮与170ml水的混合液,混合物搅拌过夜,逐渐析出黄色颗粒状固体,过滤,水洗涤,将固体加入5l反应瓶,再加入1700ml二氯甲烷和1700ml水,缓慢搅拌下加入醋酸调ph 6,过滤,固体水洗、丙酮洗,烘干得亮黄色固体266g(收率91.4%),hplc纯度98.8%。
[0068]
实施例2:4
‑
(6
‑
((6
‑
溴
‑8‑
环戊基
‑
7,8
‑
二氢
‑5‑
甲基
‑7‑
氧代吡啶并[2,3
‑
d]嘧啶
‑2‑
基)氨基)
‑3‑
吡啶基)
‑1‑
哌嗪羧酸叔丁酯(式4化合物)的实验室制备
[0069]
5l反应瓶中,加入167g(0.6mol)4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,1600ml甲苯,通氮气保护,于20~30℃滴加750ml1mol/l的六甲基二硅基氨基锂的四氢呋喃溶液,保持20~30℃搅拌反应30分钟。将171g(0.5mol)6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮分散在600ml甲苯中,缓慢倒入反应液,加毕,保持30℃反应4h,tlc检测6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮消失为反应终点(展开剂:乙酸乙酯/正己烷=1/4)。反应毕,缓慢加入340ml丙酮与170ml水的混合液,混合物搅拌过夜,逐渐析出黄色颗粒状固体,过滤,水洗涤,将固体加入5l反应瓶,再加入1700ml二氯甲烷和1700ml水,缓慢搅拌下加入醋酸调ph 6,过滤,固体水洗、丙酮洗,烘干的亮黄色固体263g(收率90.4%),hplc纯度98.9%。
[0070]
实施例3:4
‑
(6
‑
((6
‑
溴
‑8‑
环戊基
‑
7,8
‑
二氢
‑5‑
甲基
‑7‑
氧代吡啶并[2,3
‑
d]嘧啶
‑2‑
基)氨基)
‑3‑
吡啶基)
‑1‑
哌嗪羧酸叔丁酯(式4化合物)的工业化制备
[0071]
2000l反应釜中,加入725kg甲苯,84kg 4
‑
(6
‑
氨基吡啶
‑3‑
基)哌嗪
‑1‑
羧酸叔丁酯,通氮气保护,于20~30℃滴加375l 1mol/l的六甲基二硅基氨基钠的四氢呋喃溶液,保持20~30℃搅拌反应30分钟。将86kg 6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮分散在270kg甲苯中,缓慢倒入反应液,加毕,保持30℃反应4h,tlc检测6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮消失为反应终点(展开剂:乙酸乙酯/正己烷=1/4)。反应毕,缓慢加入130kg丙酮与85kg水的混合液,混合物搅拌过夜,逐渐析出黄色颗粒状固体,过滤,水洗涤,将固体加入2000l反应釜,再加入1120kg二氯甲烷和850kg水,缓慢搅拌下加入醋酸调ph 6,离心,固体水洗、丙酮洗,烘干的亮黄色固体133kg(收率90.6%),hplc纯度98.7%。
[0072]
实施例4:2
‑
甲基
‑2‑
丙基
‑4‑
(6
‑
{[8
‑
环戊基
‑5‑
甲基
‑7‑
氧代
‑6‑
(1
‑
丁氧乙烯基)
‑
7,8
‑
二氢吡啶并[2,3
‑
d]嘧啶
‑2‑
基]氨基}
‑3‑
吡啶基)
‑1‑
哌嗪甲酸(式5化合物)的实验室制备
[0073]
5l反应瓶中,加入200g(0.34mol)式4化合物,1600ml正丁醇,160ml水,51g(0.51mol)正丁基乙烯基醚及66g(0.51mol)二异丙基乙胺,通氮气保护,升温至95℃。加入0.2g双(2
‑
二苯基膦基苯基)醚及4g三苯基膦,搅拌10分钟,再加入0.04g氯化钯及2g碘化亚铜,氮气环境下,保持95℃反应20h,hplc检测原料消失为反应终点。反应毕,热过滤除去不溶物,降温至60℃,加入500ml水,搅拌分层,弃去水相,有机相减压浓缩至干,加入2000ml乙酸乙酯及4g三乙胺,加热回流至固体溶解,降温至0~10℃析晶4h,过滤,烘干得亮黄色固体196g,收率95.3%,hplc纯度99.3%。
[0074]
实施例5:2
‑
甲基
‑2‑
丙基
‑4‑
(6
‑
{[8
‑
环戊基
‑5‑
甲基
‑7‑
氧代
‑6‑
(1
‑
丁氧乙烯基)
‑
7,8
‑
二氢吡啶并[2,3
‑
d]嘧啶
‑2‑
基]氨基}
‑3‑
吡啶基)
‑1‑
哌嗪甲酸(式5化合物)的实验室制备
[0075]
5l反应瓶中,加入200g(0.34mol)式4化合物,1600ml正丁醇,160ml水,51g(0.51mol)正丁基乙烯基醚及66g(0.51mol)二异丙基乙胺,通氮气保护,升温至95℃。加入0.2g双(2
‑
二苯基膦基苯基)醚及4g三苯基膦,搅拌10分钟,再加入0.04g氯化钯及2g碘化亚铜,氮气环境下,保持95℃反应20h,hplc检测原料消失为反应终点。反应毕,热过滤除去不溶物,降温至60℃,加入500ml水,搅拌分层,弃去水相,有机相减压浓缩至干,加入2000ml醋酸异丙酯及4g n
‑
甲基吗啉,加热回流至固体溶解,降温至0~10℃析晶4h,过滤,烘干得亮黄色固体194g,收率93.9%,hplc纯度99.2%。
[0076]
实施例6:2
‑
甲基
‑2‑
丙基
‑4‑
(6
‑
{[8
‑
环戊基
‑5‑
甲基
‑7‑
氧代
‑6‑
(1
‑
丁氧乙烯基)
‑
7,8
‑
二氢吡啶并[2,3
‑
d]嘧啶
‑2‑
基]氨基}
‑3‑
吡啶基)
‑1‑
哌嗪甲酸(式5化合物)的工业化制备
[0077]
3000l反应釜中,加入200kg式4化合物,1450kg正丁醇,160kg水,51kg正丁基乙烯基醚及66kg二异丙基乙胺,通氮气保护,升温至95℃。加入0.2kg双(2
‑
二苯基膦基苯基)醚及4kg三苯基膦,搅拌10分钟,再加入40g氯化钯及2kg碘化亚铜,氮气环境下,保持95℃反应20h,hplc检测原料消失为反应终点。反应毕,趁热压滤,降温至60℃,加入500kg水,搅拌分层,弃去水相,有机相减压浓缩至干,加入1800kg醋酸异丙酯及4kg二异丙基乙胺,加热回流
至固体溶解,降温至0~10℃析晶4h,离心,烘干得亮黄色固体198kg,收率95.9%,hplc纯度99.5%。
[0078]
实施例7:帕布昔利布的实验室制备
[0079]
5000ml反应瓶中,加入100g(0.166mol)式5化合物,1000ml正丁醇,1500ml苯甲醚及1000ml水,升温至80℃,滴加50g(0.25mol,40%)氢溴酸溶液,滴毕,保温反应20h,hplc检测控制反应终点。反应毕,降温至60℃,用30%氢氧化钠溶液调ph值至10,分液,弃去水层,再用1000ml*2水洗涤2次,将上层液体升温至80℃,过滤除去不溶物,滤液减压蒸馏出约600ml溶剂,剩余物料缓慢降温至0~5℃,析晶3h,过滤,烘干得69.4g高纯度亮黄色帕布昔利布,收率93.8%,hplc纯度99.92%。
[0080]
实施例8:帕布昔利布的工业化制备
[0081]
5000l搪玻璃反应釜中,加入100kg式5化合物,900kg正丁醇,1500kg苯甲醚及1000kg水,升温至80℃,滴加25kg(0.25mol,37.3%)盐酸溶液,滴毕,保温反应20h,hplc检测控制反应终点。反应毕,降温至60℃,用30%氢氧化钠溶液调ph10,分液,弃去水层,再用1000kg*2水洗涤两次,将上层液体升温至80℃,通过0.45um滤膜(pp材质)压滤至d级区,滤液减压蒸馏出约550kg溶剂,剩余物料缓慢降温至0~5℃,析晶3h,过滤,烘干得70.1kg高纯度亮黄色帕布昔利布,收率94.7%,hplc纯度99.95%。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。