一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的合成方法
技术领域
[0001]
本发明涉及有机化工中间体合成技术领域,具体涉及一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的合成方法。
背景技术:
[0002]
吡唑并吡啶化合物是一类非常重要的稠杂环,由于其广泛的生理活性以及与吲哚、氮杂吲哚等在结构上的类似性,近年来引起了人们广泛的研究兴趣。该类化合物在防治克氏阴性和阳性细菌、肿瘤和癌症、哮喘病、神经性疾病、骨骼疏松症和老年痴呆症等方面有很好的疗效。因此人们对该类化合物的研究越来越深入和广泛,1h
‑
吡唑并[3,4
‑
b]吡啶骨架被应用于构建多种具有药物活性的分子或抑制剂,具有很强的药理学意义。3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑ꢀ
羧酸甲酯作为具有双官能团的1h
‑
吡唑并[3,4
‑
b]吡啶衍生物,有望应用于高活性片段参与杀菌药物及肿瘤抑制药物的开发。
[0003]
但在1h
‑
吡唑并[3,4
‑
b]吡啶框架上同时进行甲醛基及甲酸甲酯基的双重官能团添加,较为困难,难以实现。
[0004]
且现有技术中,3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯尚未见报道,故而对3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的制备进行研究是有必要的。
技术实现要素:
[0005]
本发明的目的在于提供了一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的合成方法,该合成方法不仅解决了具有双官能团的1h
‑
吡唑并[3,4
‑
b]吡啶衍生物合成困难的问题,而且反应条件温和,适合工业化批量生产。
[0006]
为达成上述目的,本发明采用以下技术方案:
[0007]
一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的合成方法,包括以下步骤:
[0008]
第一步:将化合物1、cdi、二甲羟胺盐酸盐加入有机溶剂中,加热反应后,经后处理纯化,得到化合物2;
[0009]
第二步:将dhp和化合物2加入有机溶剂中,再加入一水合对甲苯磺酸,搅拌反应完毕后,经后处理纯化,得到化合物3;
[0010]
第三步:将化合物3溶于有机溶剂中,加入四氢铝锂搅拌反应完毕后,经后处理纯化,得到化合物4;
[0011]
第四步:在室温条件下,将化合物4、甲醇和碱溶于有机溶剂中,氩气置换后加入钯催化剂,再经一氧化碳置换后在一氧化碳氛围下60
‑
80℃反应 16
‑
20小时,经后处理纯化,得到化合物5;
[0012]
第五步:将化合物5溶于酸溶液中,0
‑
60℃反应12
‑
20小时,经后处理纯化,即得;
[0013]
其合成路线如下:
[0014][0015]
在1h
‑
吡唑并[3,4
‑
b]吡啶衍生物的制备中,常规策略是在1h
‑
吡唑并[3,4
‑
b] 吡啶框架上进行基团的添加,然而本技术所述3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑ꢀ
羧酸甲酯具有双官能团,若采用在1h
‑
吡唑并[3,4
‑
b]吡啶框架上直接进行甲醛基及甲酸甲酯基的添加,较为困难。为获得本技术所述目标化合物,发明人进行了合成策略的调整,采用具有吡唑并[3,4
‑
b]吡啶结构,且具有3位羧酸取代基及 5位卤素取代基的5
‑
溴
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑3‑
羧酸作为原料,通过羧酸至 weinreb酰胺的转换以及环上氨基的保护,进行酰胺还原获得3
‑
位醛基化,实现了羧酸到醛基的官能团转换;在卤素至甲酸甲酯官能团转化过程中,通常可以采取多种策略,例如将卤代中间体制备成格式试剂进行反应,然而格式试剂在反应过程中,需严格除水除氧,且该卤代中间体自身含有醛基,也会与格式试剂进行反应,故而该方法不仅操作上存在诸多不便,反应副产物也较多。本技术采用钯催化剂催化下,一氧化碳气氛中进行插羰反应,完成卤素至甲酸甲酯官能团的转化;反应过程中,卤代中间体与钯进行氧化加成生成钯配合物,体系中的一氧化碳与钯进行配位,再进一步进行羰基的转移插入,形成酰钯化合物,该酰钯化合物与溶剂进行配体交换,发生还原消除,最终得到目标化合物。上述合成路线中,所用原料价廉易得,合成步骤简单,操作简便,反应条件温和,未采用超低温超高压等严苛反应条件,尤其是合成步骤四中,采用过渡金属催化剂进行反应,反应过程清洁,便于后处理;同时,反应具有单一性,副产物少,易于纯化,适合工艺放大,具有较高的经济效益。
[0016]
优选的,所述第一步中,加热至65℃反应3
‑
5小时。
[0017]
优选的,所述第二步始终在室温下进行反应,搅拌反应时间为16
‑
20小时。
[0018]
优选的,所述第三步是先降温至0℃,然后分批加入四氢铝锂,在0℃条件下搅拌1
‑
3小时。
[0019]
优选的,第四步所述有机溶剂可以是dmf、dma、dmso、dmac中的一种或多种。
[0020]
优选的,第四步所述碱为有机碱或者无机碱。
[0021]
优选的,有机碱为二乙胺、三乙胺、仲丁胺、二异丙基乙胺或二氮杂二环中的一种或多种。
[0022]
优选的,无机碱为碳酸钾、醋酸钾、碳酸钠中的一种或多种。
[0023]
优选的,第四步所述钯催化剂为pd(dppf)cl2、pdcl2(ph3p)2、pd(ph3p)4、 pd2(dba)3、pd(oac)2中的一种或多种。
[0024]
优选的,第四步所述钯催化剂为pd2(dba)3或/和pd(oac)2时,需与膦配体配合使用。
[0025]
优选的,第四步所述膦配体为dppp、dppf、ph3p、dppe中的一种或多种。
[0026]
优选的,所述第四步中,化合物4与钯催化剂的摩尔比为1:0.05~0.2。
[0027]
优选的,所述第四步中,化合物4与碱的摩尔比为1:2.5~5。
[0028]
优选的,所述第四步中,使用的催化剂为pd(dppf)cl2,在co氛围下80℃下搅拌16小时。
[0029]
优选的,所述第四步具体过程为:室温氩气保护下,将甲醇和三乙胺依次加入到化合物4的dmf溶液中;氩气置换下,加入pd(dppf)cl2,再经co置换后在co氛围下80℃下搅拌16小时;反应完毕后,将反应液加入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品过柱纯化得到化合物5。
[0030]
优选的,所述第五步中,使用的酸溶液为三氟乙酸的二氯甲烷溶液或者hcl 的1,4
‑
二氧六环溶液或者hcl的甲醇溶液。
[0031]
优选的,所述第五步中,反应时间为16小时。
[0032]
优选的,所述第五步具体过程为:室温下,将化合物5加入到三氟乙酸的二氯甲烷溶液中,反应液在室温下搅拌16小时;反应完毕后,将反应液加入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品过柱纯化得到3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯。
[0033]
有益效果:
[0034]
本发明提出了一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的制备方法,为双官能团的1h
‑
吡唑并[3,4
‑
b]吡啶衍生物的制备提供了可靠的参考,并且该化合物可应用于高活性片段参与杀菌药物及肿瘤抑制药物的研究,应用于更高效的杀菌药物及肿瘤抑制剂的制备。
[0035]
本发明在制备过程中,反应条件温和,未采用超低温超高压等严苛反应条件,尤其是合成步骤4中,采用过渡金属催化剂进行反应,反应过程清洁,便于后处理;同时,反应具有单一性,副产物少,易于纯化,适合工艺放大,具有较高的经济效益。
[0036]
应当理解,前述构思以及在下面更加详细地描述的额外构思的所有组合只要在这样的构思不相互矛盾的情况下都可以被视为本公开的发明主题的一部分。
[0037]
结合附图从下面的描述中可以更加全面地理解本发明教导的前述和其他方面、实施例和特征。本发明的其他附加方面例如示例性实施方式的特征和/或有益效果将在下面的描述中显见,或通过根据本发明教导的具体实施方式的实践中得知。
附图说明
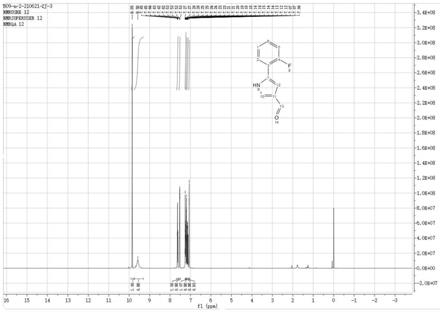
[0038]
附图不意在按比例绘制。在附图中,在各个图中示出的每个相同或近似相同的组成部分可以用相同的标号表示。为了清晰起见,在每个图中,并非每个组成部分均被标记。现在,将通过例子并参考附图来描述本发明的各个方面的实施例,其中:
[0039]
图1为3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的核磁氢谱图。
具体实施方式
[0040]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例的附图,对本发明实施例的技术方案进行清楚、完整地描述。显然,所描述的实施例是本发
明的一部分实施例,而不是全部的实施例。基于所描述的本发明的实施例,本领域普通技术人员在无需创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。除非另作定义,此处使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。
[0041]
本发明先以5
‑
溴
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑3‑
羧酸为原料,经过缩合得到weinreb酰胺5
‑
溴
‑
n
‑
甲氧基
‑
n
‑
甲基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑3‑
羧酰胺。再经 dhp保护得到5
‑
溴
‑
n
‑
甲氧基
‑
n
‑
甲基
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
1h
‑
吡唑并 [3,4
‑
b]吡啶
‑3‑
甲酰胺。weinreb酰胺用四氢铝锂还原得到5
‑
溴
‑1‑
(四氢
‑
2h
‑ꢀ
吡喃
‑2‑
基)
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑3‑
甲醛。以5
‑
溴
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑ꢀ
基)
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑3‑
甲醛经过插羰反应得到3
‑
甲酰基
‑1‑
(四氢
‑
2h
‑ꢀ
吡喃
‑2‑
基)
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯,最后通过脱thp保护得到产品3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯。
[0042]
所述3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的合成方法,具体包括以下步骤:
[0043]
第一步(1):将化合物1、cdi、二甲羟胺盐酸盐加入有机溶剂中,加热反应后,纯化,得到化合物2;
[0044]
第二步(2):将dhp和化合物2加入有机溶剂中,再加入一水合对甲苯磺酸,搅拌反应完毕后,经后处理纯化,得到化合物3;
[0045]
第三步(3):将化合物3溶于有机溶剂中,加入四氢铝锂搅拌反应完毕后,经后处理纯化,得到化合物4;
[0046]
第四步(4):在室温条件下,将化合物4、甲醇和碱溶于有机溶剂中,氩气置换后加入钯催化剂,再经一氧化碳置换后在一氧化碳氛围下60
‑
80℃反应16
‑
20小时,经后处理纯化,得到化合物5;
[0047]
第五步(5):将化合物5溶于酸溶液中,0
‑
60℃反应12
‑
20小时,经后处理纯化,即得。
[0048]
所述第一步到第三步制备得到化合物4的过程,不限于上述方法,上述方法仅仅是本发明列举的一种优选的制备方法。
[0049]
具体的,所述第一步中,加热至65℃反应3
‑
5小时。此时化合物2的收率为87.1%。
[0050]
具体的,所述第二步始终在室温下进行反应,搅拌反应时间为16
‑
20小时。此时化合物3的收率为92.2%。
[0051]
具体的,所述第三步是先降温至0℃,然后分批加入四氢铝锂,在0℃条件下搅拌1
‑
3小时。此时化合物4的收率为62.1%。
[0052]
应当指出的是,现有技术里记载的第三步的收率能够达到90%以上,但是发明人根据其方法,并不能达到90%的收率,甚至不到60%;因此,本发明的发明人在现有技术的基础上,对反应条件进行了优化,最终使第三步的收率有所提高,达到了62.1%。
[0053]
上述步骤中,所述第四步中,有机溶剂可以是dmf、dma、dmso、 dmac中的一种或多种。
[0054]
所述第四步中,碱为有机碱或者无机碱。
[0055]
具体的,有机碱为二乙胺、三乙胺、仲丁胺、二异丙基乙胺或二氮杂二环中的一种或多种。
[0056]
具体的,无机碱为碳酸钾、醋酸钾、碳酸钠中的一种或多种。
[0057]
所述第四步中,所用的钯催化剂为pd(dppf)cl2、pdcl2(ph3p)2、 pd(ph3p)4、pd2(dba)3、pd(oac)2中的一种或多种。
[0058]
具体的,所述钯催化剂为pd2(dba)3或/和pd(oac)2时,还使用了膦配体。因为钯催化剂为pd2(dba)3或/和pd(oac)2时,需与膦配体共同作用。
[0059]
具体的,所述的膦配体为dppp、dppf、ph3p、dppe中的一种或多种。
[0060]
所述第四步中,化合物4与钯催化剂的摩尔比为1:0.05~0.2。
[0061]
所述第四步中,化合物4与碱的摩尔比为1:2.5~5。
[0062]
所述第四步中,使用钯催化剂pd(dppf)cl2,在co氛围下80℃下搅拌16小时。这样的反应条件可以大大提高本发明的收率。
[0063]
具体的,所述第四步具体过程为:室温氩气保护下,将甲醇和三乙胺依次加入到化合物4的dmf溶液中;体系氩气置换下,加入pd(dppf)cl2,再经co 置换后在co氛围下80℃下搅拌16小时;反应完毕后,将反应液加入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品过柱纯化得到化合物5。
[0064]
所述第五步中,使用的酸溶液为三氟乙酸的二氯甲烷溶液、hcl的1,4
‑
二氧六环溶液或者hcl的甲醇溶液。
[0065]
具体的,所述第五步具体过程为:室温下,将化合物5加入到三氟乙酸的二氯甲烷溶液中,反应液在室温下搅拌16小时;反应完毕后,将反应液加入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品过柱纯化得到3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯。
[0066]
值得一提的是,本发明所用的条件多为常温常压反应,如第四步的co氛围下也无需加压,在常温条件下即可得到本发明所需产物;即使是唯一需要加热的第四步,其加热条件也不超过80℃。这对于工业化的生产很有必要。
[0067]
本发明实施例中所用的原料和试剂均市售可得,在本技术文件中,“室温条件下”是指20
‑
25℃温度范围。
[0068]
通过下述实施例将有助于理解本发明,但不限制于本发明的内容。
[0069]
实施例1
[0070]
一种3
‑
甲酰基
‑1‑
(四氢
‑
2h
‑
吡喃
‑2‑
基)
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的制备
[0071]
向250ml单口瓶中依次加入dmf 70ml,甲醇40ml和三乙胺(7.8g, 5.0eq)、化合物4(4.8g,1.0eq);体系氩气置换下,将pd(dppf)cl2(1.1g, 0.1eq)加入。经co置换后,反应液在co氛围下80℃下搅拌16小时。反应完毕后,将反应液倒入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品淋柱纯化得到3.86g化合物5。
[0072]
收率:86.7%。hplc:97.3%。
[0073]1h nmr(400mhz,cdcl3)δ10.24(s,1h),9.25(dd,j=17.4,1.8hz,2 h),6.29(dd,j=10.3,2.5hz,1h),4.17(d,j=8.7hz,1h),4.00(s,3h),3.88 (m,j=11.4hz,1h),2.63(m,j=8.0hz,1h),2.20(m,1h),2.05(m,j=14. 0hz,1h),1.85(m,j=8.8hz,3h)。
[0074]
m/z(ei):290.1(m h)
。
[0075]
一种3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的制备
[0076]
室温下,将化合物5(3g,1.0eq)加入到4n的tfa/dcm(30ml)溶液中,反应液在室温(20
‑
25℃)下搅拌16小时。反应完毕后,将反应液加入水中,用乙酸乙酯萃取,有机相合并,饱和食盐水洗,干燥后浓缩得到粗品;粗品经柱层析纯化得到1.82g产物。
[0077]
图1所示为3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯的核磁氢谱图。
[0078]
收率:85.5%,hplc:98.3%。
[0079]1hnmr(400mhz,dmso)δ15.08(s,1h),10.19(s,1h),9.16(d,j=2.1hz, 1h),8.98(d,j=2.0hz,1h),3.94(s,3h)。
[0080]
m/z(ei):206.0(m h)
。
[0081]
实施例2
‑
9的步骤与实施例1相同,仅改变了步骤(4)的部分工艺条件。为使结果更加直观明了,实施例2
‑
9采用了列表的方式,对本发明进行了具体说明。
[0082]
另外,本发明还提供了对比例1
‑
6,其步骤亦如上。
[0083]
具体如表1所示,实施例和对比例均选取1.0eq化合物4。
[0084]
表1 步骤(4)中各反应条件对产物收率的影响
[0085]
[0086][0087]
实施例1
‑
9,在0.1eqpdcl2(ph3p)2或0.1eq pd(oac)2‑
dppf或0.1eq pd(dppf)cl2催化体系中,分别采用三乙胺、二异丙基乙胺、醋酸钾、碳酸钾作碱,于80℃下反应16小时实施本发明,反应结果较为理想,所得产物收率均在70%以上;反应中,催化剂pdcl2(ph3p)2、pd(oac)2–
dppf、pd(dppf)cl2经还原得到pd(0), pd(0)先与化合物4进行氧化加成,形成钯配合物,启动反应,而后进行一氧化碳的转移插入,以及与溶剂的配体交换,得到化合物5;上述催化剂均表现出较优的反应活性,其中pd(dppf)cl2能够更好地实现本发明;此外,反应中分别采用三乙胺、二异丙基乙胺、醋酸钾、碳酸钾中和反应过程中生成的氢溴酸副产物,从而促进反应的正向进行,其中5.0eq的三乙胺效果最优。
[0088]
对比例1
‑
5相较于实施例1
‑
9,分别采用不同用量的催化剂、不同用量的碱以及不同反应温度、不同反应时长进行反应,所得反应结果均劣于实施例1
‑
9。具体为:对比例1较实施例1
‑
9,提高反应温度至90℃,反应温度促使主反应进行的同时,在很大程度上也促进了副反应的发生,使产物收率降低;对比例2 较实施例1
‑
9,降低反应温度至50℃,反应温度的降低,减缓了反应进行的速率,使同等反应时间下的反应收率大幅度降低;对比例3
‑
4较实施例1
‑
9,改变了催化剂的用量,对比例3将催化剂用量增加至0.3eq,催化剂用量的增多促进副反应进行的同时,也进一步增加反应的生产成本;而对比例4将催化剂用量降低至0.03eq,这无疑将延缓反应进程,使反应收率降低明显;对比例5较实施例 1
‑
9增加了碱的用量,对比例6较实施例1
‑
9延长了反应时间,二者均大幅度促进了副反应的发生,使反应收率降低。
[0089]
实施例10
‑
15的步骤与实施例1相同,仅改变了步骤(5)的部分工艺条件。为使结果更加直观明了,实施例10
‑
15采用了列表的方式,对本发明进行了具体说明。
[0090]
另外,本发明还提供了对比例7
‑
9,其步骤亦如上。
[0091]
具体如表2所示,实施例和对比例均选取1.0eq化合物5。
[0092]
表2 步骤(5)中各反应条件对产物收率的影响
[0093]
序号酸溶液反应温度反应时间/h目标化合物收率实施例104n hcl/dioxane室温1680.3%实施例114n hcl/meoh室温1677.4%实施例124n tfa/dcm室温1278.0%实施例134n tfa/dcm室温2081.0%实施例144n tfa/dcm0℃1667.3%实施例154n tfa/dcm60℃1673.2%对比例74n tfa/dcm80℃1660.3%对比例84n tfa/dcm室温554.6%对比例94n tfa/dcm室温3066.8%
[0094]
实施例1及实施例10
‑
15中,化合物5分别在4n hcl/dioxane、4n hcl/meoh 以及4n tfa/dcm中,于不同反应温度、不同时长内实施本发明,脱保护得到目标化合物3
‑
甲酰基
‑
1h
‑
吡唑并[3,4
‑
b]吡啶
‑5‑
羧酸甲酯。由于所用酸溶液为强酸溶液,且thp保护基团较易离去,反应温度不宜过高,优选为0
‑
60℃,并进一步优选为室温(20
‑
25℃);对比例7较实施例1及实施例10
‑
15,将反应温度升高至80℃,化合物5所带基团易发生副反应的同时,操作上也存在不便;在反应时长上,对比例8较实施例1及实施例10
‑
15缩短反应时间至5h,该反应时间内,化合物5未能反应完全,产物收率降低明显;对比例9较实施例1 及实施例10
‑
15延长反应时间至30h,反应时间的延长,增加了反应所需能耗,促进了副反应的发生,产物收率降低,故而反应时长优选12
‑
20h;进一步获得优选实施例1。
[0095]
本发明反应条件温和,合成步骤精简,反应过程清洁,后处理简单,对环境友好,操作简便,副产物少,纯度高,收率高,具有良好的应用前景。
[0096]
虽然本发明已以较佳实施例揭露如上,然其并非用以限定本发明。本发明所属技术领域中具有通常知识者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰。因此,本发明的保护范围当视权利要求书所界定者为准。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
![一种3-甲酰基-1H-吡唑并[3,4-b]吡啶-5-羧酸甲酯的合成方法与流程](http://img.xjishu.com/img/zl/2021/11/16/qqsf8glub.jpg)