1.本发明涉及粗对苯二甲酸加氢精制催化剂及其制备方法。
背景技术:
2.精对苯二甲酸,俗称pta,是合成聚对苯二甲酸乙二醇酯(pet)的基本原料。负载型钯/炭催化剂适用于粗对苯二甲酸的精制,粗对苯二甲酸中的对羧基苯甲醛(简称4-cba)等杂质进行加氢转变为其它的化合物后,随后采用结晶的方法来分离提纯。由于钯/炭催化剂采用单一的活性组份,所以金属钯在载体上的分布状况,对催化剂性能的影响非常大。
3.对苯二甲酸加氢精制通常反应压力为6.5~8.5mpa,反应温度为250~290℃条件下进行,由于对苯二甲酸加氢精制反应过程是一个一级反应,反应速度快,反应过程中反应物难以穿透到催化剂颗粒的内部进行反应,这就使得颗粒内部的活性金属由于位阻影响,接触不到直径较大的反应物分子组份不能发挥作用。此时,外表面的活性金属表现出的高的催化活性。出于充分利用贵金属的考虑,通常钯/炭催化剂做成蛋壳型,即让活性组份钯主要负载于载体的表面。钯与反应物接触的表面积越大,活性也越好。蛋壳型活性组份分布的催化剂比分布范围较宽的催化剂具有更高的加氢催化能力。但反应初期由于钯反应活性过高,导致出现对苯二甲酸发生过加氢而生成对甲基苯甲酸(p-ta),p-ta在水中的溶解度比对甲羟基苯甲酸(hmba)和苯甲酸(ba)的小很多,因此反应产品中过多的p-ta难除去,这样会导致pta产品中p-ta超标。美国专利us4,892,972(purification of crude terephthalic acid)通过采用pd/c和rh/c的双层催化剂,pd与rh的比例为10:1用于粗对苯二甲酸加氢精制,从而减少pta产品中的对甲基苯甲酸(p-ta)含量,但rh的价格是pd的十倍,因此在实际中不适用。
技术实现要素:
4.本发明所要解决的技术问题之一是现有技术中催化剂初活性高导致加氢精制产品中对甲基苯甲酸偏高的问题,提供一种新的用于粗对苯二甲酸加氢精制催化剂,该催化剂用于粗对苯二甲酸的加氢精制反应,具有对甲基苯甲酸低的特点。
5.本发明所要解决的技术问题之二是与上述技术问题之一相对应的催化剂的制备方法。
6.本发明所要解决的问题之三是上述催化剂的应用。
7.本发明所要解决的问题之四是粗对苯二甲酸加氢精制的方法。
8.为解决上述技术问题之一,本发明采用的技术方案如下:
9.粗对苯二甲酸加氢精制催化剂,所述催化剂包括载体和活性组分,所述的载体为活性炭,所述活性组分为钯元素,所述钯元素包括pd0和pd
2
,且pd
2
与pd0的重量比为0.3~3。
10.我们惊奇地发现,当催化剂中所述钯元素包括pd0和pd
2
,且pd
2
与pd0的重量比为0.3~3时,该催化剂用于粗对苯二甲酸的加氢精制反应,具有对甲基苯甲酸残留量低的优
点。
11.上述技术方案中,作为非限制性举例,pd
2
与pd0的重量比可以为0.35、0.4、0.45、0.5、0.55、0.60、0.65、0.70、0.75、0.80、0.85、0.9、0.95、1.0等等,更优选0.4~0.7。
12.上述技术方案中,催化剂中钯元素含量优选为0.2~1.0wt%。例如但不限于0.25wt%、0.30wt%、0.35wt%、0.4wt%、0.45wt%、0.5wt%、0.55wt%、0.60wt%、0.65wt%、0.70wt%等等。
13.上述技术方案中,在催化剂中,优选pd
2
以pdo的形式存在。
14.上述技术方案中,所述的活性炭优选为煤质炭、木质炭或果壳炭。
15.上述技术方案中,所述的果壳炭优选为椰壳炭。
16.上述技术方案中,所述载体的比表面优选为800~1600m2/g。例如但不限于850m2/g、900m2/g、950m2/g、1000m2/g、1500m2/g、2000m2/g等等。
17.上述技术方案中,所述载体的孔容优选为0.35~0.80ml/g。例如但不限于0.40ml/g、0.45ml/g、0.50ml/g、0.55ml/g、0.60ml/g、0.65ml/g、0.70ml/g、0.75ml/g等等。
18.为解决上述技术问题之二,本发明采用的技术方案如下:
19.上述技术问题之一的技术方案所述的催化剂的制备方法,包括以下步骤:
20.(1)通过碱性化合物调节含钯化合物水溶液的ph值至1~10,得到催化剂前驱体;
21.(2)将载体与催化剂前驱体混合,得到催化剂前体i;
22.(3)将步骤(2)中的到催化剂前体i陈化,得到催化剂前体ii;
23.(4)将步骤(3)的催化剂前体ii在惰性气氛下热处理使得钯元素以pdo形式存在,得到催化剂前体iii;
24.(5)用还原剂将步骤(4)所述催化剂前体iii中的部分化合态钯还原为pd0,得到催化剂前体iv;
25.(6)水洗除去催化剂前体iv中的杂质,得到所述的催化剂。
26.上述技术方案中,步骤(2)所述的催化剂载体可以直接使用市售活性炭,还可以在步骤(1)之前增加:
27.(i)将市售活性炭水洗、烘干;和/或
28.(ii)将市售活性炭在含氧化剂的水溶液中处理,然后将沥干、烘干。
29.上述技术方案中,步骤(4)热处理的温度优选300~600℃,例如但不限于350℃、400℃、450℃、500℃、550℃等等。
30.上述技术方案中,步骤(4)热处理的时间优选为1~8h,例如但不限于1.5h、2h、2.5h、3h、3.5h、4h、4.5h、5h、5.5h、6h、6.5h、7h、7.5h等等;
31.上述技术方案中,步骤(1)所述碱性化合物优选为碱金属氢氧化物、碱金属碳酸盐或氨中的至少一种,最优选为碱金属碳酸盐,碳酸钠最常见,最便宜,因此最最优选碳酸钠,碳酸钠水溶液的浓度优选为5~15wt%(例如但不限6wt%、7wt%、8wt%、9wt%、10wt%、11wt%、12wt%、13wt%、14wt%)。
32.上述技术方案中,步骤(1)所述的含钯化合物选自硝酸钯、醋酸钯、氯钯酸及其盐和二氯四氨合钯中至少一种,优选为氯钯酸。
33.上述技术方案中,步骤(1)所述的ph值更优选为3~7,例如但不限3.5、4.0、4.5、5.0、5.5、6.0、6.5。
1486.6ev),工作电压10kv,x射线电流20ma采用污染碳c1s(eb=284.6ev)作能量校正,此时条件下,335.2ev(pd3d
5/2
)和340.6ev(pd3d
3/2
)为pd0所对应的特征峰,337.1ev(pd3d
5/2
)和342.2ev(pd3d
3/2
)为pd
2
所对应的特征峰,采用xps peakfit4.1软件对pd3d峰进行拟合分峰然后进行计算。
50.采用xps测定不同价态钯的百分含量,计算公式如下:
[0051][0052]
x:被分析价态的pd;i:光电子峰面积;n:被考虑的pd中不同价态的个数;s:灵敏因子
[0053]
本发明方法得到的催化剂采用高压釜进行评价,具体评价条件为:
[0054]
催化剂用量:2.0克;粗对苯二甲酸量(cta):30.0克;溶剂:1000g纯水;反应压力:7.5mpa;反应温度:280℃;反应时间45min。
[0055]
高效液相色谱(hplc)分析反应前后溶液中的4-cba,p-ta,hmba(对甲羟基苯甲酸)、ba(苯甲酸)。
[0056]
pd含量采用icp进行分析。
[0057]
下面通过实施例和附图说明对本发明作进一步阐述。
附图说明
[0058]
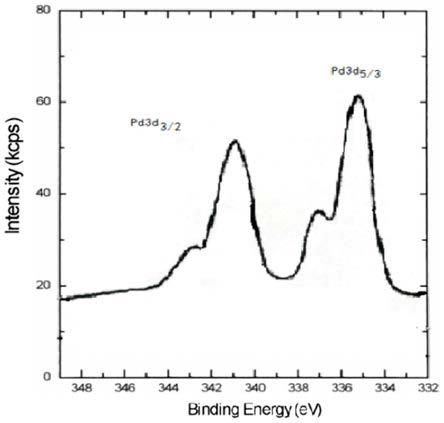
图1为实施例1中钯炭催化剂中pd0和pd
2
在3d区xps图谱。
具体实施方式
[0059]
【实施例1】
[0060]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0061]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体;
[0062]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0063]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0064]
催化剂前体i在重量空速为50h-1
n2气氛下350℃处理4h,冷却至25℃,得到催化剂前体ii。
[0065]
在25℃下,将催化剂前体ii浸渍在20克10wt%的乙醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。
[0066]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0067]
采用xps测定不同价态钯的百分含量,催化剂的xps图谱如图1所示。
[0068]
icp测催化剂中pd的质量百分含量。
[0069]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0070]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0071]
【实施例2】
[0072]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0073]
将烘干后的活性炭在含4wt%hno3和4wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体。
[0074]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0075]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0076]
催化剂前体i在重量空速为50h-1
n2气氛下350℃处理4h,冷却至25℃,得到催化剂前体ii。
[0077]
在25℃下,将催化剂前体ii浸渍在20克10wt%的乙醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。
[0078]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0079]
采用xps测定不同价态钯的百分含量。
[0080]
icp测催化剂中pd的质量百分含量。
[0081]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0082]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0083]
【实施例3】
[0084]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0085]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体。
[0086]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0087]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0088]
催化剂前体i在重量空速为50h-1
n2气氛下450℃处理4h,冷却至25℃,得到催化剂前体ii。
[0089]
在25℃下,将催化剂前体ii浸渍在20克10wt%的乙醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。
[0090]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0091]
采用xps测定不同价态钯的百分含量。
[0092]
icp测催化剂中pd的质量百分含量。
[0093]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0094]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0095]
【实施例4】
[0096]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0097]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体。
[0098]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0099]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0100]
催化剂前体i在重量空速为50h-1
n2气氛下350℃处理4h,冷却至25℃,得到催化剂前体ii。
[0101]
在25℃下,将催化剂前体ii浸渍在80克10wt%的乙醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0102]
采用xps测定不同价态钯的百分含量。
[0103]
icp测催化剂中pd的质量百分含量。
[0104]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0105]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0106]
【对比例1】
[0107]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0108]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体;
[0109]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0110]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0111]
在25℃下,将催化剂前体i浸渍在20克10wt%的乙醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体ii。
[0112]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0113]
采用xps测定不同价态钯的百分含量。
[0114]
icp测催化剂中pd的质量百分含量。
[0115]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的
杂质。
[0116]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0117]
对比例1与实施例1比较发现,由于缺少像实施例1那样的催化剂前体i热处理步骤,未将化合态钯转化为pbo形式,从而即使在还原步骤仍保留有pd
2
,在最终产品中pd
2
也几乎不存在,导致pd
2
水洗流失,致使催化剂加氢活性低,产品中4-cba和p-ta含量都很高,无法得到本发明所述的催化剂。
[0118]
【对比例2】
[0119]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0120]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体;
[0121]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0122]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0123]
催化剂前体i在重量空速为50h-1
n2气氛下350℃处理4h,冷却至25℃,得到催化剂前体ii。
[0124]
在25℃下,将催化剂前体ii浸渍在7.0克10wt%的甲醛水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。
[0125]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0126]
采用xps测定不同价态钯的百分含量,催化剂的xps图谱如图1所示。
[0127]
icp测催化剂中pd的质量百分含量。
[0128]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0129]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0130]
对比例2与实施例1比较发现,甲醛水溶液还原性比乙醛水溶液的还原性强,有容易将pd
2
全部还原成pd0的弊端,导致产品中的p-ta含量高。
[0131]
【对比例3】
[0132]
用甲酸钠代替实施例1中的乙醛作为还原剂还原剂,还原剂的氧化还原当量数用量相同,具体为:
[0133]
称取50克市售的4~8目、片状椰壳活性炭(比表面为1100m2/g,孔容为0.52ml/g)用纯水洗涤,纯水与活性炭体积之比为5:1,然后沥干、烘干。
[0134]
将烘干后的活性炭在含2wt%hno3和2wt%h2o2水溶液中25℃浸泡1.5h,水溶液与活性炭体积之比为5:1,然后将沥干、烘干,得到催化剂载体。
[0135]
催化剂前驱体的配制:称取1.25克含钯20wt%的氯钯酸水溶液,用10wt%的碳酸钠水溶液边搅拌边滴加调节氯钯酸水溶液ph至5.0,然后加纯水定容至26ml,搅拌均匀得到催化剂前驱体。
[0136]
将上述催化剂载体浸渍在催化剂前驱体中,陈化24小时,得到催化剂前体i。
[0137]
催化剂前体i在重量空速为50h-1
n2气氛下350℃处理4h,冷却至25℃,得到催化剂前体ii。
[0138]
在25℃下,将催化剂前体ii浸渍在31克10wt%的甲酸钠水溶液中进行还原,还原时间2h,还原后得到催化剂前体iii。
[0139]
用纯水洗涤催化剂前体iii至洗涤液用agno3检测无cl-为止,干燥得到所需催化剂。
[0140]
采用xps测定不同价态钯的百分含量。
[0141]
icp测催化剂中pd的质量百分含量。
[0142]
采用高压釜对催化剂进行评价,采用高效液相色谱(hplc)分析反应前后溶液中的杂质。
[0143]
为便于比较,将催化剂的分析数据和评价结果数据分别列于表1和表2。
[0144]
对比例3与实施例1同比发现,甲酸钠水溶液还原性比乙醛水溶液的还原性强,有容易将pd
2
全部还原成pd0的弊端,导致产品中的p-ta含量高。
[0145]
表1
[0146][0147]
表2
[0148][0149]
备注:原料cta中杂质含量4-cba:3025ppmw;p-ta:768ppmw;hmba:56ppmw;ba:38ppmw。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。