制备
α
‑
烯丙基化环烷酮的方法
技术领域
1.本发明涉及一种通式(iii)所表示的α
‑
烯丙基化环烷酮的制造方法。
背景技术:
2.已知大环状化合物在医药品、香料、农药等领域中显示有用的活性。作为大环状酮的一种的麝香烯酮是可生物降解性优异、具有高残香性和优雅的质感的香料原材料。为了应对近年来的易分解性的合成麝香原材料的需求的提高,要求开发安全且高效率的制造方法。
[0003][0004]
麝香烯酮可以通过包括将环十二酮的α位烯丙基化,得到2
‑
(2
‑
甲基烯丙基)环十二酮的工序;进一步由2
‑
(2
‑
甲基烯丙基)环十二酮进行转化的多个工序的方法而得到。作为环十二酮的α位烯丙基化,报道有以下的方法。使环十二烷在对甲苯磺酸存在下与甲醇反应,然后,使用填充柱分离1,1
‑
二甲氧基环十二酮。也报道了在丙酸存在下使巴豆醇与该1,1
‑
二甲氧基环十二酮反应来制造2
‑
(1
‑
甲基芳基)环十二酮的方法(专利文献1)。
[0005][0006]
此外,报道了虽然不是大环状化合物,但在环状酮的α位导入烯丙基的反应(非专利文献1)。使环己酮在对甲苯磺酸一水合物的存在下与烯丙醇反应,其后,通过减压蒸馏,分离二烯丙醇缩环己酮。接着,将二烯丙醇缩环己酮在对甲苯磺酸存在下进行加热,制造2
‑
烯丙基环己酮(非专利文献1)。根据该方法,收率为85~91%左右。
[0007][0008]
现有技术文献
[0009]
专利文献
[0010]
专利文献1:日本特表2015
‑
533799号
[0011]
非专利文献
[0012]
非专利文献1:w.l.howard,n.b.lorette,organic synthesis,vol.5,1973,p.292
技术实现要素:
[0013]
发明所要解决的课题
[0014]
如上所述,将环烷酮作为起始物质,为了制造α
‑
烯丙基化环烷酮,已知在各种酸的存在下进行反应。然而,未知以提高的收率得到高纯度的α
‑
烯丙基化环烷酮的方法。
[0015]
本发明的课题在于提供一种以环状化合物的环烷酮为起始物质、以提高的收率得到高纯度的α
‑
烯丙基化环烷酮的方法。
[0016]
用于解决课题的技术手段
[0017]
本发明人等惊讶地发现,使环烷酮在酸催化剂存在下与醇进行反应,接着即使没有分离和精制工序,通过在酸催化剂存在下与乙烯基醇反应,也能够以提高的收率且高纯度得到α
‑
烯丙基化环烷酮。
[0018]
即,本发明是制造通式(iii)所表示的α
‑
烯丙基化环烷酮(以下,有时称为“式(iii)的化合物”或“化合物(iii)”)的方法,其中,包括:
[0019]
工序1:使通式(i)所表示的化合物(环烷酮)(以下,有时称为“式(i)的化合物”或“化合物(i)”)以及碳原子数为1以上且4以下的醇,在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序;以及
[0020]
工序2:使工序1中得到的粗产物与通式(ii)所表示的化合物(以下,有时称为“式(ii)的化合物”或“化合物(ii)”)在第二酸催化剂的存在下进行反应,得到通式(iii)所表示的α
‑
烯丙基化环烷酮的工序,
[0021]
连续地进行所述工序1和工序2。
[0022][0023]
所述式中,
[0024]
基团
‑
a1‑
(其中,前面的结合键是指与碳原子c1结合的结合键,后面的结合键是指与碳原子c2结合的结合键)是可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基,
[0025]
r4为氢原子或碳原子数为1以上且4以下的烷基。
[0026]
发明的效果
[0027]
根据本发明,可以以环状化合物的环烷酮为起始物质,以提高的收率、且提高了高纯度的产物的收率得到α
‑
烯丙基化环烷酮。
具体实施方式
[0028]
在本技术说明书中,“麝香烯酮”是指firmenich sa(geneva,瑞士)的香料,是各种异构体的外消旋混合物。具体而言,麝香烯酮主要是z
‑3‑
甲基
‑
环十五碳
‑5‑
烯
‑1‑
酮、e
‑3‑
甲基
‑
环十五碳
‑5‑
烯
‑1‑
酮、e
‑3‑
甲基
‑
环十五碳
‑4‑
烯
‑1‑
酮和z
‑3‑
甲基
‑
环十五碳
‑4‑
烯
‑1‑
酮的混合物的总称。
[0029]
<式(i)的化合物、式(ii)的化合物、式(iii)的化合物、式(xx)的化合物、式(xxi)的化合物>
[0030]
在所述式(i)的化合物、式(iii)的化合物、式(xx)的化合物以及式(xxi)的化合物中,基团
‑
a1‑
的“可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基”的“碳原子数为4以上且20以下的亚烷基”例如以基团
‑
(ch2)4‑
、基团
‑
(ch2)5‑
、基团
‑
(ch2)6‑
、基团
‑
(ch2)7‑
、基团
‑
(ch2)8‑
、基团
‑
(ch2)9‑
、基团
‑
(ch2)
10
‑
、基团
‑
(ch2)
11
‑
、基团
‑
(ch2)
12
‑
、基团
‑
(ch2)
13
‑
、基团
‑
(ch2)
14
‑
、基团
‑
(ch2)
15
‑
、基团
‑
(ch2)
16
‑
、基团
‑
(ch2)
17
‑
、基团
‑
(ch2)
18
‑
、基团
‑
(ch2)
19
‑
和基团
‑
(ch2)
20
‑
表示。从将得到的通式(iii)的化合物用作香料化合物的前体的观点和/或式(iii)的化合物合成时(工序1和工序2)的温度变高的观点出发,作为所述“碳原子数为4以上且20以下的亚烷基”,优选碳原子数为6以上且14以下的亚烷基,更优选碳原子数为8以上且14以下,进一步优选碳原子数为10以上且14以下,进一步更优选碳原子数为10以上且12以下的亚烷基。
[0031]
所述式(i)的化合物、式(iii)的化合物、式(xx)的化合物以及式(xxi)的化合物的中,基团
‑
a1‑
的“可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基”的“可以任选地包含杂原子的碳原子数为4以上且20以下的亚烷基”可以包含氧、氮和/或硫原子作为杂原子。即,“可以任选地包含杂原子的碳原子数为4以上且20以下的亚烷基”是可以包含不阻碍反应的醚键(
‑
o
‑
)、酯键(
‑
c(=o)
‑
o
‑
或
‑
o
‑
c(=o)
‑
)、硫醚基(
‑
s
‑
)或这些的碳原子数为4以上且20以下的亚烷基。所述“可以进一步包含醚键、酯键、硫醚基或这些的碳原子数为4以上且20以下的亚烷基”例如可以举出基团
‑
(ch2)2‑
o
‑
(ch2)2‑
、基团
‑
(ch2)2‑
o
‑
(ch2)6‑
、基团
‑
(ch2)3‑
o
‑
(ch2)5‑
、基团
‑
(ch2)4‑
o
‑
(ch2)4‑
、基团
‑
(ch2)2‑
o
‑
(ch2)7‑
、基团
‑
(ch2)3‑
o
‑
(ch2)6‑
、基团
‑
(ch2)4‑
o
‑
(ch2)5‑
、基团
‑
(ch2)
‑
o
‑
(ch2)9‑
、基团
‑
(ch2)2‑
o
‑
(ch2)8‑
、基团
‑
(ch2)3‑
o
‑
(ch2)7‑
、基团
‑
(ch2)4‑
o
‑
(ch2)6‑
、基团
‑
(ch2)5‑
o
‑
(ch2)5‑
、基团
‑
(ch2)2‑
nh
‑
(ch2)2‑
等,从将得到的通式(iii)的化合物用作香料化合物的前体的观点出发,优选为基团
‑
(ch2)
‑
o
‑
(ch2)9‑
、基团
‑
(ch2)2‑
o
‑
(ch2)8‑
、基团
‑
(ch2)3‑
o
‑
(ch2)7‑
、基团
‑
(ch2)4‑
o
‑
(ch2)6‑
、基团
‑
(ch2)5‑
o
‑
(ch2)5‑
。
[0032]
基团
‑
a1‑
的“可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上20以下的亚烷基”为可以具有1个以上、优选为1个或2个取代基的“可以任选地包含杂原子的碳原子数为4以上且20以下的亚烷基”。作为所述取代基,可以举出烷基、烷氧基、烷基氨基、烷氧基羰基、烷酰基、芳基、芳烷基、芳氧基、酰氧基、羧基、卤素原子、碳环、杂环等,优选烷基、烷氧基羰基、烷氧基,更优选烷基。需要说明的是,在所述取代基为烷基的情况下,该烷基的碳原子数不包含在“可以任选地包含杂原子的碳原子数为4以上且20以下的亚烷基”的碳原子数4以上且20以下。
[0033]
所述取代基的2个以上也可以相互键合而与所述取代基所键合的原子一起形成碳环或杂环。
[0034]
所述式(ii)的化合物以及式(iii)的化合物中,r4为氢原子或碳原子数为1以上且4以下的烷基。此外,作为r4,从有助于克莱森重排时的稳定的构象形成,其结果提高通式(iii)的化合物的收率的观点出发,优选为氢原子或碳原子数为1以上且3以下的烷基,更优
选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0035]
所述通式(i)所表示的化合物例如以以下的式子表示,从将得到的通式(iii)的化合物用作香料化合物的前体的观点出发,优选式(vi)所表示的化合物、式(vii)所表示的化合物、式(viii)所表示的化合物、式(ix)所表示的化合物,更优选式(vii)所表示的化合物、式(viii)所表示的化合物。式(vii)所表示的化合物为环十二酮。需要说明的是,式(vii)所表示的化合物为后述的式(i
‑
1)的化合物。
[0036][0037]
所述通式(i)所表示的化合物可以通过市售获得,或者通过公知的方法,例如日本特开2016
‑
34937中记载的方法获得。
[0038]
所述通式(ii)所表示的化合物例如以以下的式子表示,从将所得到的通式(iii)的化合物用作香料化合物的前体的观点出发,优选为以下的式(62)所表示的β
‑
甲基烯丙醇。所述通式(ii)所表示的化合物可以通过市售获得,或者通过公知的方法,例如日本特开2002
‑
105010中记载的方法获得。需要说明的是,式(62)所表示的化合物为后述的式(ii
‑
1)的化合物。
[0039][0040]
所述通式(iii)所表示的化合物例如以以下的式子表示,从将得到的通式(iii)的化合物用作香料化合物的前体的观点出发,优选式(xxvi)所表示的化合物、式(xxvii)所表示的化合物、式(xxviii)所表示的化合物、式(xxix)所表示的化合物,更优选式(xxvii)所表示的化合物、式(xxviii)所表示的化合物。式(xxvii)所表示的化合物是2
‑
(2
‑
甲基烯丙基)环十二酮。需要说明的是,式(xxvii)所表示的化合物为后述的式(iii
‑
1)的化合物。
[0041][0042]
[制备通式(iii)所表示的α
‑
烯丙基化环烷酮的方法]
[0043]
<工序1:使通式(i)所表示的化合物以及碳原子数为1以上且4以下的醇,在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序>
[0044]
<醇>
[0045]
在本发明中,所述醇是碳原子数为1以上且4以下的醇。作为所述醇,优选为碳原子数为1以上且4以下的饱和醇。作为所述醇,可以举出甲醇、乙醇、1
‑
丙醇、1
‑
丁醇、2
‑
甲基丙醇等。
[0046]
<第一酸催化剂>
[0047]
在本发明中,所述第一酸催化剂可以为选自有机磺酸或其盐以及吡啶的无机酸盐中的1种以上。所述有机磺酸例如为芳香族磺酸或脂肪族磺酸,优选为芳香族磺酸。此外,所述有机磺酸的盐可以是吡啶鎓盐。
[0048]
所述芳香族磺酸或其盐可以举出苯系芳香族化合物或其盐、杂芳香族化合物或其盐。
[0049]
苯系芳香族化合物或其盐可以是具有1个苯环的化合物或其盐。具有1个苯环的化合物,具体而言,可以举出:苯磺酸、对甲苯磺酸、4
‑
氨基苯磺酸、2
‑
氨基苯磺酸、1h
‑
苯并咪唑
‑2‑
磺酸、氟苯磺酸、二氟苯磺酸、三氟苯磺酸、氯苯磺酸、二氯苯磺酸、三氯苯磺酸。作为具有1个苯环的化合物,优选苯磺酸、对甲苯磺酸、氟苯磺酸、二氟苯磺酸、三氟苯磺酸、氯苯磺酸、二氯苯磺酸、三氯苯磺酸,更优选对甲苯磺酸。
[0050]
作为所述具有1个苯环的化合物的盐,具体而言,可以举出2,4,6
‑
三甲基吡啶鎓对甲苯磺酸盐。
[0051]
所述苯系芳香族化合物或其盐可以是具有2个以上苯环的化合物或其盐,具体而言,可以举出萘磺酸、蒽磺酸。
[0052]
所述杂芳香族化合物或其盐可以是包含氮原子的化合物。具体而言,所述杂芳香族化合物或其盐可以举出5
‑
异喹啉磺酸、8
‑
喹啉磺酸、6
‑
喹啉磺酸、4
‑
吡啶乙磺酸、2
‑
吡啶乙磺酸、3
‑
吡啶磺酸。
[0053]
对于所述脂肪族磺酸或其盐,作为苯系芳香族化合物或其盐、杂芳香族化合物或其盐以外的化合物,具体而言,可以举出甲磺酸、三氟甲磺酸、10
‑
樟脑磺酸、4
‑
吗啉丙磺酸等或其盐。作为所述脂肪族磺酸或其盐,优选三氟甲磺酸或10
‑
樟脑磺酸,更优选10
‑
樟脑磺酸。
[0054]
作为构成所述吡啶的无机酸盐的酸,具体而言,可以是盐酸、硫酸、硝酸、磷酸、亚硫酸、亚硝酸、氢溴酸、氢碘酸。其中,特别优选盐酸、硫酸、硝酸或磷酸,更优选盐酸。
[0055]
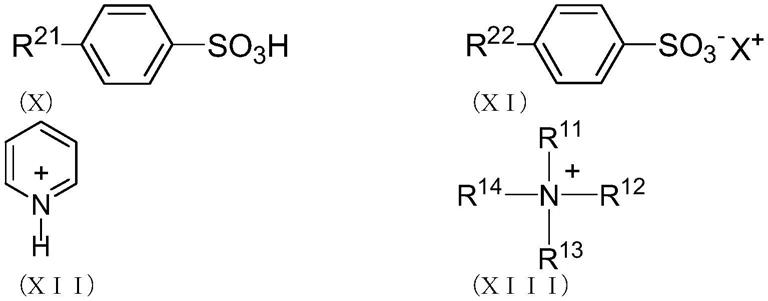
在本发明中,所述第一酸催化剂可以选自以下的式(x)所表示的化合物和式(xi)所表示的化合物。
[0056][0057]
所述式中,r
21
和r
22
各自独立地为氢原子或碳原子数为1以上且5以下的烷基,x
以式(xii)或式(xiii)表示,
[0058]
所述式中,r
11
、r
12
、r
13
以及r
14
相同或不同,为氢原子或碳原子数为1以上且5以下的烷基。
[0059]
所述式(xiii)中,从收率良好地制造得到的通式(iii)的化合物的观点出发,r
11
、r
12
、r
13
和r
14
优选为氢原子或碳原子数为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0060]
所述式(x)中,从即使在高温条件下也不挥发而稳定地不引起通式(i)的化合物的异构化,且促进克莱森重排的观点出发,r
21
优选为氢原子或碳原子数为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0061]
所述式(xi)中,从是比较弱的酸,即使在高温条件下也不挥发而稳定地不引起通式(ii)的化合物的异构化、且促进克莱森重排的观点出发,r
22
优选为氢原子或碳原子数为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0062]
所述第一酸催化剂由于促进式(i)的化合物的缩酮化,因此,可以举出对甲苯磺酸、对甲苯磺酸吡啶鎓等,优选对甲苯磺酸或对甲苯磺酸吡啶鎓。
[0063]
在本发明中,所述第一酸催化剂的使用量相对于所述式(i)的化合物优选为10
‑5当量以上、更优选为10
‑4当量以上、进一步优选为5
×
10
‑4当量以上,且优选为1当量以下、更优选为0.5当量以下、进一步优选为0.2当量以下。这是因为,如果第一酸催化剂的使用量为上述范围,则作为所述第一酸催化剂可以促进式(i)的化合物的缩酮化。
[0064]
<脱水剂>
[0065]
所述脱水剂例如可以举出原羧酸酯或硫酸盐。具体而言,所述脱水剂可以举出:原甲酸三甲酯、原甲酸三乙酯、na2so4、mgso4等。所述脱水剂优选为原羧酸酯,更优选为原羧酸
三甲酯或原羧酸三甲酯,更优选为原甲酸三甲酯。如果使用所述脱水剂,则促进工序1的反应,因此优选。因此,工序1优选在所述第一酸催化剂与所述脱水剂的存在下进行。
[0066]
<反应温度>
[0067]
在本发明中,使式(i)的化合物以及碳原子数为1以上且4以下的醇在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序例如在120℃以上、优选为125℃以上、更优选为135℃以上,例如在150℃以下、优选为145℃以下进行。这是因为,如果在该范围内进行该工序,则会促进式(i)的化合物的缩酮化。
[0068]
<反应时间>
[0069]
在本发明中,使式(i)的化合物和碳原子数为1以上且4以下的醇在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序的反应时间例如为2小时~5天,优选为4小时~2天,从生产成本和生产效率的观点出发,更优选为6小时~24小时。
[0070]
<反应装置>
[0071]
本发明中,工序1以及后述的工序2优选使用精馏塔进行。
[0072]
在本发明中,工序1和后述的工序2优选以一锅法进行。本发明中,一锅法是指不经过将成为中间体的化合物蒸馏除去到体系外的操作,而在同一容器内进行2个以上的反应。
[0073]
需要说明的是,在本发明中,工序1和工序2由于连续地进行且不进行分离,因此,通过工序1得到以下的式(xx)的化合物和/或式(xxi)的化合物。r1oh、r2oh和/或r3oh与工序1中的碳原子数为1以上且4以下的醇对应。
[0074][0075]
所述式中,r1、r2和r3相同或不同,为碳原子数为1以上且4以下的烷基,
[0076]
基团
‑
a1‑
(其中,前面的结合键是指与碳原子c1结合的结合键,后面的结合键是指与碳原子c2结合的结合键),是可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基。
[0077]
<工序2:使工序1中得到的粗产物与通式(ii)所表示的化合物在第二酸催化剂的存在下进行反应,得到通式(iii)所表示的α
‑
烯丙基化环烷酮的工序>
[0078]
<第二酸催化剂>
[0079]
在本发明中,所述第二酸催化剂可以为选自有机磺酸或其盐以及吡啶的无机酸盐中的1种以上。所述有机磺酸例如为芳香族磺酸或脂肪族磺酸,优选为芳香族磺酸。此外,所述有机磺酸的盐可以是吡啶鎓盐。
[0080]
所述芳香族磺酸或其盐可以举出苯系芳香族化合物或其盐、杂芳香族化合物或其盐。
[0081]
苯系芳香族化合物或其盐可以是具有1个苯环的化合物或其盐。具有1个苯环的化
合物具体而言,可以举出:苯磺酸、对甲苯磺酸、4
‑
氨基苯磺酸、2
‑
氨基苯磺酸、1h
‑
苯并咪唑
‑2‑
磺酸、氟苯磺酸、二氟苯磺酸、三氟苯磺酸、氯苯磺酸、二氯苯磺酸、三氯苯磺酸。作为具有1个苯环的化合物,优选苯磺酸、对甲苯磺酸、氟苯磺酸、二氟苯磺酸、三氟苯磺酸、氯苯磺酸、二氯苯磺酸、三氯苯磺酸,更优选对甲苯磺酸。
[0082]
作为所述具有1个苯环的化合物的盐,具体而言,可以举出2,4,6
‑
三甲基吡啶鎓对甲苯磺酸盐。
[0083]
所述苯系芳香族化合物或其盐可以是具有2个以上苯环的化合物或其盐,具体而言,可以举出萘磺酸、蒽磺酸。
[0084]
所述杂芳香族化合物或其盐可以是包含氮原子的化合物。具体而言,所述杂芳香族化合物或其盐可以举出5
‑
异喹啉磺酸、8
‑
喹啉磺酸、6
‑
喹啉磺酸、4
‑
吡啶乙磺酸、2
‑
吡啶乙磺酸、3
‑
吡啶磺酸。
[0085]
对于所述脂肪族磺酸或其盐,作为苯系芳香族化合物或其盐、杂芳香族化合物或其盐以外的化合物,具体而言,可以举出甲磺酸、三氟甲磺酸、10
‑
樟脑磺酸、4
‑
吗啉丙磺酸等或其盐。作为所述脂肪族磺酸或其盐,优选三氟甲磺酸或10
‑
樟脑磺酸,更优选10
‑
樟脑磺酸。
[0086]
作为构成所述吡啶的无机酸盐的酸,具体而言,可以为盐酸、硫酸、硝酸、磷酸、亚硫酸、亚硝酸、氢溴酸、氢碘酸。其中,特别优选盐酸、硫酸、硝酸或磷酸,更优选盐酸。
[0087]
在本发明中,所述第二酸催化剂可以选自以下的式(x)所表示的化合物和式(xi)所表示的化合物。
[0088][0089]
所述式中,r
21
和r
22
各自独立地为氢原子或碳原子数为1以上且5以下的烷基,x
以式(xii)或式(xiii)表示,
[0090]
所述式中,r
11
、r
12
、r
13
以及r
14
相同或不同,为氢原子或碳原子数为1以上且5以下的烷基。
[0091]
所述式(xiii)中,从收率良好地制造得到的通式(iii)的化合物的观点出发,r
11
、r
12
、r
13
和r
14
优选为氢原子或碳原子数为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0092]
所述式(x)中,从即使在高温条件下也不挥发而稳定地不引起通式(ii)的化合物的异构化、且促进克莱森重排的观点出发,r
21
优选为氢原子或碳原子数为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0093]
所述式(xi)中,从是比较弱的酸,即使在高温条件下也不挥发而稳定地不引起通式(ii)的化合物的异构化,并且促进克莱森重排的观点出发,r
22
优选为氢原子或碳原子数
为1以上且3以下的烷基,更优选为氢原子、
‑
ch3或
‑
c2h5,进一步更优选为
‑
ch3。
[0094]
所述第二酸催化剂可以举出对甲苯磺酸、对甲苯磺酸吡啶鎓等,优选对甲苯磺酸或对甲苯磺酸吡啶鎓。
[0095]
在本发明中,所述第二酸催化剂的使用量相对于所述式(i)的化合物优选为10
‑5当量以上、更优选为10
‑4当量以上、进一步优选为5
×
10
‑4当量以上,且优选为1当量以下、更优选为0.5当量以下、进一步优选为0.2当量以下。这是因为,如果第二酸催化剂的使用量为上述范围,则作为所述第二酸催化剂,可以促进式(i)的化合物的缩酮化。
[0096]
在本发明中,所述第二酸催化剂可以与工序1中的第一酸催化剂相同或不同。本发明更优选工序1中的第一酸催化剂与工序2中的第二酸催化剂相同。本发明更优选可以在工序1之后实质上不追加催化剂而进行工序2。在该情况下,使用第一酸催化剂作为第二酸催化剂。
[0097]
<连续>
[0098]
本发明是连续地进行所述工序1和工序2的方法。在本发明中,连续是指不需要在工序1与工序2之间进行操作,并不排除放置或储藏。如果连续地进行所述工序1和工序2,则具有能够无中途的分离和精制工序地进行的效果。连续地进行的方法优选不具有中途的分离和精制工序。连续地进行所述工序1和工序2时,例如在香料的合成中不需要除去反应副产物,在这一点上,本发明的方法优异。
[0099]
<分离和精制工序>
[0100]
在本发明中,“分离和精制工序”是指“在下一工序中对作为原料的化合物进行分级的操作”。具体而言,是指“进行蒸馏而在下一工序中将作为原料的化合物蒸馏除去至体系外进行分级”、或“利用硅胶柱色谱法在下一工序中对作为原料的化合物进行分级”。在本发明中,从反应混合物中通过蒸馏和水洗除去溶剂或残存的试剂的操作不包含在分离和精制工序中。例如,在压力50kpa以上进行蒸馏,在下一工序中不对作为原料的化合物进行分级的情况下,不包含在本技术的分离和精制工序中。例如,在压力5kpa以下进行蒸馏,在下一工序中对作为原料的化合物进行分级的情况下,相当于本技术的分离和精制工序。
[0101]
<反应温度>
[0102]
在本发明中,工序2例如在120℃以上、优选为125℃以上、更优选为135℃以上,且例如150℃以下、优选为145℃以下进行。这是因为,如果在该范围内进行该工序,则能够使在通式(xx)所表示的化合物和/或通式(xxi)所表示的化合物与通式(ii)所表示的化合物之间进行缩醛交换的结果中产生的醇(r1oh、r2oh、r3oh)向反应体系外挥发,使反应加速。
[0103]
<反应时间>
[0104]
在本发明中,工序2的反应时间例如为2小时~5天,优选为4小时~2天,从生产成本和生产效率的观点出发,更优选为6小时~24小时。
[0105]
此外,本发明是制造通式(iii)所表示的α
‑
烯丙基化环烷酮的方法,其中,包括:
[0106]
工序1:使通式(i)所表示的化合物以及碳原子数为1以上且4以下的醇,在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序;以及
[0107]
工序2:使工序1中得到的粗产物与通式(ii)所表示的化合物在第二酸催化剂的存在下进行反应,得到通式(iii)所表示的α
‑
烯丙基化环烷酮的工序,
[0108]
所述工序1的第一酸催化剂与工序2的第二酸催化剂相同。
[0109][0110]
[所述式中,
[0111]
基团
‑
a1‑
(其中,前面的结合键是指与碳原子c1结合的结合键,后面的结合键是指与碳原子c2结合的结合键)是可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基,
[0112]
r4为氢原子或碳原子数为1以上且4以下的烷基。]
[0113]
在本发明的方法中,优选所述式(i)为下述式(i
‑
1),所述式(iii)为下述式(iii
‑
1)。这是因为,在后述的麝香烯酮的合成方法中可以得到有用的式(iii
‑
1)的化合物。
[0114][0115]
此外,本发明是麝香烯酮的合成方法,其中,使用了通过上述方法制造的式(iii
‑
1)的α
‑
烯丙基化环烷酮。此外,本发明是作为麝香烯酮的原料的式(iii
‑
1)的α
‑
烯丙基化环烷酮的用途。
[0116][0117]
所述麝香烯酮的合成方法具体而言包括以下的工序:
[0118]
(i)式(iii
‑
1)的化合物的环化、
[0119]
(ii)氢化、
[0120]
(iii)氧化裂解、
[0121]
(iv)还原、以及
[0122]
(v)开环。
[0123][0124]
关于上述的实施方式,本发明还公开以下的方法。
[0125]
<1>一种制造通式(iii)所表示的α
‑
烯丙基化环烷酮的方法,其中,包括:
[0126]
工序1:使通式(i)所表示的化合物以及碳原子数为1以上且4以下的醇,在第一酸催化剂以及根据情况的脱水剂的存在下进行反应的工序;以及
[0127]
工序2:使工序1中得到的粗产物与通式(ii)所表示的化合物在第二酸催化剂的存在下进行反应,得到通式(iii)所表示的α
‑
烯丙基化环烷酮的工序,
[0128]
连续地进行所述工序1和工序2。
[0129][0130]
[所述中,基团
‑
a1‑
(其中,前面的结合键是指与碳原子c1结合的结合键,后面的结合键是指与碳原子c2结合的结合键)是可以任选地包含杂原子,并且可以任选地具有取代基的碳原子数为4以上且20以下的亚烷基,r4为氢原子或碳原子数为1以上且4以下的烷基。]
[0131]
<2>如<1>所述的方法,其中,在所述第一酸催化剂和所述脱水剂的存在下进行工序1。
[0132]
<3>根据<1>或<2>所述的方法,其中,连续地进行的方法不具有中途的分离和精制工序。
[0133]
<4>如<1>~<3>中任一项所述的方法,其中,所述第一酸催化剂和所述第二酸催化剂各自独立地为选自有机磺酸或其盐以及吡啶的无机酸盐中的1种以上。
[0134]
<5>如<4>所述的方法,其中,所述有机磺酸为芳香族磺酸。
[0135]
<6>根据<4>或<5>所述的方法,其中,所述第一酸催化剂和所述第二酸催化剂各自独立地选自以下的式(x)所表示的化合物和式(xi)所表示的化合物。
[0136][0137]
[所述式中,r
21
和r
22
各自独立地为氢原子或碳原子数为1以上且5以下的烷基,x
以式(xii)或式(xiii)表示,
[0138]
所述式中,r
11
、r
12
、r
13
以及r
14
相同或不同,为氢原子或碳原子数为1以上且5以下的烷基。]
[0139]
<7>如<1>~<6>中任一项所述的方法,其中,所述第一酸催化剂和所述第二酸催化剂各自独立地包含对甲苯磺酸或对甲苯磺酸吡啶鎓。
[0140]
<8>如<4>~<6>中任一项所述的方法,其中,所述有机磺酸的盐为吡啶鎓盐。
[0141]
<9>如<4>所述的方法,其中,构成所述吡啶的无机酸盐的酸为选自盐酸、硫酸、硝酸、磷酸、亚硫酸、亚硝酸、氢溴酸、氢碘酸、乙酸以及丁酸中的1种以上。
[0142]
<10>如<1>~<9>中任一项所述的方法,其中,所述第一酸催化剂与所述第二酸催化剂相同。
[0143]
<11>如<1>~<10>中任一项所述的方法,其中,所述第一酸催化剂的使用量相对于所述通式(i)的化合物和通式(ii)的化合物的合计为10
‑5当量以上且1当量以下。
[0144]
<12>如<1>~<11>中任一项所述的方法,其中,在120℃以上且145℃以下进行第一酸催化剂的存在下的反应。
[0145]
<13>如<1>至<12>中任一项所述的方法,其中,在120℃以上且145℃以下进行第二酸催化剂的存在下的反应。
[0146]
<14>如<1>~<13>中任一项所述的方法,其中,所述第一工序和所述第二工序使用精馏塔进行。
[0147]
<15>如<1>~<14>中任一项所述的方法,其中,所述第一工序和所述第二工序以一锅法进行。
[0148]
<16>如<1>~<15>中任一项所述的方法,其中,所述式(i)为下述式(i
‑
1),所述式(iii)为下述式(iii
‑
1)。
[0149][0150]
<17>一种麝香烯酮的合成方法,其中,使用了通过<16>所述的方法制造的式(iii
‑
1)的α
‑
烯丙基化环烷酮。
[0151]
[实施例]
[0152]
<气相色谱法(gc)的装置以及分析条件>
[0153]
gc装置:agilent technologies inc.制造、型号:gc
‑
6850
[0154]
柱:j&w公司制造、db
‑
1(内径0.25mm、长度30m、膜厚0.25μm)
[0155]
载气:he,1.5ml/min
[0156]
注入条件:300℃、分流比100/1
[0157]
注入量:1μl
[0158]
检测条件:fid方式、300℃
[0159]
柱温度条件:80℃
→
10℃/分钟升温
→
300℃保持10分钟
[0160]
[化合物的鉴定]
[0161]
在以下的实施例、实验例等中得到的各化合物通过gc(气相色谱法)鉴定。
[0162]
收率(%)通过以下的数学式算出。
[0163][0164]
在此,gc面积%表示该成分被gc检测并输出的图表面积相对于整体的比例。
[0165]
[实施例1]2
‑
(2
‑
甲基烯丙基)环十二酮的合成
[0166][0167]
(i)工序1
[0168]
在2l四口烧瓶中加入环十二酮(i
‑
1)(500.0g、2.743mol)、原甲酸三甲酯(349.5g、3.292mol)和甲醇(264.3g、8.229mol),在室温下搅拌,制成均匀溶液。向其中加入对甲苯磺酸吡啶鎓(ppts、0.7g、2.743mmol)并进行搅拌,使其溶解。在2l四口烧瓶安装温度计、机械搅拌器和10级贯通式精馏塔(共和化学公司制造)。在氮气气氛下,在外部温度80℃下开始2l四口烧瓶中的内容物的搅拌。从2l四口烧瓶中经时地对反应物进行取样,通过gc分析观察(i
‑
1)的化合物的转化率。从反应开始6小时时停止反应,进行冷却。
[0169]
接着,进行产物中所含的甲醇和原甲酸三甲酯的减压蒸馏除去。在2l四口烧瓶上安装k字管、冷却管以及馏分接收器。在氮气气氛下,在110℃的外部温度下开始从产物中的减压蒸馏除去。开始1小时后,从常压减压至66.5kpa,继续减压蒸馏除去。在2小时停止向接收器的馏出,结束减压蒸馏除去。对蒸馏除去后的反应液进行气相色谱分析,结果成分组成为:1,1
‑
二甲氧基环十二烷(xx
‑
1)为43.5gc面积%、1
‑
甲氧基
‑1‑
环十二烯(xxi
‑
1)为54.5gc面积%。
[0170][0171]
(ii)工序2
[0172]
在装有上述反应物的2l四口烧瓶上安装温度计、机械搅拌器和10级贯通式精馏塔。在室温下,将β
‑
甲基烯丙醇(ii
‑
1)(296.7g、4.115mol)添加到上述四口烧瓶中。对烧瓶内容物(包含对甲苯磺酸吡啶鎓(ppts、0.7g、2.743mmol))进行搅拌,制成均匀体系后,一边使氮气流动,一边使油浴的温度为140℃对外部温度进行加热。经时的确认中,监测到顶温为65℃。3.5小时后,确认上述烧瓶内容物中的1,1
‑
二甲氧基环十二烷(xx
‑
1)以及1
‑
甲氧基
‑1‑
环十二烯(xxi
‑
1)消失,停止反应。
[0173]
接着,在包含反应液(包含2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1))的2l四口烧瓶上安装k字管、冷却管以及馏分接收器,在18.0kpa、120℃下进行加热搅拌,馏出β
‑
甲基烯丙醇(ii
‑
1)(馏出量:67.1g)。加热搅拌2小时后,作为完成蒸馏,减压至16kpa,继续加热搅拌1小时。
[0174]
将蒸馏除去了β
‑
甲基烯丙醇(ii
‑
1)后的残留物转移到2l带夹套的分离式反应容器中,向其中添加将k2hpo4(0.4g、2.057mmol)溶解于离子交换水20.2g而得到的碱性水。在上述分离式反应容器中设置机械搅拌器、温度计、蛇形冷凝器(dimroth condenser)和氮气流。将分离式反应容器中的混合物在室温下搅拌1小时。搅拌结束后,用冷凝器将上述混合物加热至80℃后,静置直至分层。从上述分离式反应容器中去除水层(15.4g),确认残留物的ph,结果为8.0(ph试纸)。
[0175]
为了从上述残留物中蒸馏除去残存的β
‑
甲基烯丙醇(ii
‑
1)和水,进行了单蒸馏操作。向装有上述残留物的分离式反应容器安装k字管、冷却管和馏分接收器,在0.3kpa、130℃下加热搅拌2小时,馏出β
‑
甲基烯丙醇(ii
‑
1)和水,得到残留物(671.2g)。对上述残留物进行气相色谱分析,结果2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)为94.7gc面积%。根据得到的产量,2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的收率为98.1%。
[0176]
[实施例2]2
‑
(2
‑
甲基烯丙基)环十二酮的合成
[0177][0178]
(i)工序1
[0179]
在2l四口烧瓶中加入环十二酮(i
‑
1)(500.0g、2.743mol)、原甲酸三甲酯(349.3g、3.292mol)和甲醇(263.7g、8.229mol),进行氮气置换后,在氮气气氛下、室温下搅拌4小时,制成均匀溶液。向其中加入对甲苯磺酸吡啶鎓(ppts、0.7g、2.743mmol)并进行搅拌,使其溶解。在2l四口烧瓶上安装蛇形冷凝器,使用循环器将37℃的温水在蛇形冷凝器流动。在上述
蛇形冷凝器的前端安装dean
‑
stark型脱水管,在上述脱水管的下方安装200ml的馏分接收器。馏分接收器浸渍在冰水中进行冰冷。在dean
‑
stark型脱水管之上进一步安装蛇形冷凝器,使用另一循环器,使10℃的冷水在该蛇形冷凝器中流动。从冷却至10℃的蛇形冷凝器的上方连接硅胶管并引导至乙醇
‑
干冰阱,将其前端氮气密封。在氮气气氛下,在浴温80℃下将2l四口烧瓶中的内容物加热回流8小时。
[0180]
在装有上述反应结束物的2l四口烧瓶上,安装k字管、冷却管以及馏分接收器。在氮气气氛下,在101.3kpa下一边将浴温从100℃升温至120℃,一边用4.5小时从上述反应结束物中蒸馏除去溶剂。对蒸馏除去后的反应液进行气相色谱分析,结果成分组成为:1,1
‑
二甲氧基环十二烷(xx
‑
1)为25.9gc面积%、1
‑
甲氧基
‑1‑
环十二烯(xxi
‑
1)为73.7gc面积%、环十二酮(i
‑
1)为0.3gc面积%。
[0181]
(ii)工序2
[0182]
在装有上述反应物的2l四口烧瓶上,安装k字管、冷却管以及馏分接收器。在氮气气氛下,在浴温110℃下进行加热搅拌,向上述四口烧瓶中用8分钟滴加β
‑
甲基烯丙醇(ii
‑
1)(296.7g、4.115mol)。一边向上述四口烧瓶中流通氮气,一边在浴温110℃下蒸馏除去甲醇,直至甲基烯丙基环十二酮的含有率为40~50gc面积%。经过4小时后,停止从上述四口烧瓶中蒸馏除去甲醇(馏出量:108.9g)。从上述四口烧瓶中卸下k字管、冷却管以及馏分接收器,在上述四口烧瓶上安装蛇形冷凝器,升温至浴温130℃,将反应混合物加热回流17小时。
[0183]
接着,在装有反应液的2l四口烧瓶上安装k字管、冷却管以及馏分接收器,在18.0kpa、浴温度120℃下加热搅拌3.5小时,馏出β
‑
甲基烯丙醇(iii
‑
1)(馏出量:79.7g)。用气相色谱仪对蒸馏除去后的反应液进行分析,结果成分组成为2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)92.5面积%。
[0184]
在加入有β
‑
甲基烯丙醇(ii
‑
1)蒸馏除去后的反应液的2l四口烧瓶中,添加将k2hpo4(0.358g、2.057mmol)溶解于离子交换水20.0g而得到的碱性水,在室温下剧烈搅拌1分钟。接着,加热至浴温80℃,降低反应液的粘度,静置15分钟使其分层。上述反应液的水层的ph为8.0(ph试纸)。对上述反应液的油层进行气相色谱分析,结果2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)为92.5面积%。上述油层的产量为644.9g(理论产量:648.5g)。2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的根据得到的产量的收率为92.4%。
[0185]
[实施例3]2
‑
(2
‑
甲基烯丙基)环十二酮的合成
[0186][0187]
(i)工序1
[0188]
在带分支的反应容器(eyela制造、)上安装机械搅拌器、蛇形冷凝管。加入环十二酮(i
‑
1)(4.4g、24.1mmol)、原甲酸三乙酯(8.6g、58.2mmol)、乙醇(6.6g、140.4mmol)、对甲苯磺酸吡啶鎓(0.035g、0.13mmol),一边搅拌一边在浴温85℃下加热回流36小时。
[0189]
反应冷却后,在反应混合物中加入β
‑
甲基烯丙醇(ii
‑
1)(5.2g、72.0mmol)。安装dean
‑
stark分水器,升温至浴温140℃,加热回流11小时。
[0190]
反应冷却后,向反应溶液中加入饱和碳酸氢钠水溶液(10ml),搅拌5分钟。将所得到的反应结束物的油层用乙醚稀释,取出水层。减压蒸馏除去油层的溶剂,得到反应终液(7.0g)。反应结束液的2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的gc面积%为79.2%,收率为92.4%。
[0191][0192]
(ii)工序(ii)
[0193]
在装有上述反应物的300ml四口烧瓶上,安装k字管、冷却管以及馏分接收器。在氮气气氛下,在浴温110℃下加热搅拌,向上述四口烧瓶中用2分钟滴加β
‑
甲基烯丙醇(ii
‑
1)(29.7g、0.411mol)。一边向上述四口烧瓶中流通氮气,一边用3小时将乙醇馏出(馏出量:11.54g)。停止乙醇的馏出,从四口烧瓶卸下k字管、冷却管以及馏分接收器,向上述四口烧瓶安装蛇形冷凝器,升温至浴温130℃,将反应混合物加热回流4小时。用气相色谱仪对得到的反应液进行分析,结果成分组成为2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)为93.9面积%。需要说明的是,根据gc面积%的2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的收率为99.2%。
[0194]
将上述实施例的内容一并示于以下的表1中。
[0195]
[表1]
[0196][0197]
*1:原料是指上述通式(i)的化合物。
[0198]
根据上述表1可以理解,根据本发明的方法,能够由式(i)的化合物以提高的收率得到高纯度的式(iii)的化合物。
[0199]
实施例4~7通过同样的操作步骤求出2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的收率。以下示出实施例4的代表性的操作步骤,其他示出添加的试剂量。
[0200]
[实施例4]
[0201]
[0202]
在带分支的反应容器(eyela制造、)上安装机械搅拌器、蛇形冷凝管。加入环十二酮(i
‑
1)(4.4g、24.1mmol)、原甲酸三甲酯(3.0g、28.7mmol)、甲醇(2.3g、72.4mmol)、对甲苯磺酸(0.022g、0.12mmol),一边搅拌一边在浴温80℃下加热回流3小时。
[0203]
反应冷却后,在反应混合物中加入β
‑
甲基烯丙醇(ii
‑
1)(2.6g、36.0mmol)。安装dean
‑
stark分水器,升温至浴温140℃,加热回流2小时。
[0204]
反应冷却后,向反应溶液中加入饱和碳酸氢钠水溶液(10ml),搅拌5分钟。将所得到的反应结束物的油层用乙醚稀释,取出水层。减压蒸馏除去油层的溶剂,得到反应结束液(5.8g)。反应结束液的2
‑
[0205]
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的gc面积%为93.8%,收率为95.5%。
[0206]
[实施例5]
[0207]
除了如下变更试剂量以外,以与实施例4相同的方式进行。
[0208]
环十二酮(i
‑
1)(4.4g、24.1mmol)、原甲酸三甲酯(3.0g、28.7mmol)、甲醇(2.3g、72.4mmol)、对甲苯磺酸(0.005g、0.027mmol)、β
‑
甲基烯丙醇(ii
‑
1)(2.6g、36.0mmol),反应结束液的重量为5.4g,2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的gc面积%为95.8%,收率为90.9%。
[0209]
[实施例6]
[0210]
除了如下变更试剂量以外,以与实施例4相同的方式进行。
[0211]
环十二酮(i
‑
1)(4.4g、24.1mmol)、原甲酸三甲酯(3.0g、28.7mmol)、甲醇(2.3g、72.4mmol)、( )
‑
10
‑
樟脑磺酸(0.06g、0.25mmol)、β
‑
甲基烯丙醇(ii
‑
1)(2.6g、36.0mmol),反应结束液的重量为5.9g,2
‑
[0212]
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的gc面积%为95.3%,收率为90.9%。
[0213]
[实施例7]
[0214]
除了如下变更试剂量以外,以与实施例4相同的方式进行。
[0215]
环十二酮(i
‑
1)(4.4g、24.1mmol)、原甲酸三甲酯(3.0g、28.7mmol)、甲醇(2.3g、72.4mmol)、吡啶盐酸盐(0.26g、2.5mmol)、β
‑
甲基烯丙醇(ii
‑
1)(2.6g、36.0mmol),反应结束液的重量为5.4g,2
‑
(2
‑
甲基烯丙基)环十二酮(iii
‑
1)的gc面积%为77.1%,收率为73.7%。
[0216]
将上述实施例4~7的内容和得到的结果一并示于以下的表2和表3中。
[0217]
[表2]
[0218][0219]
*1:原料是指上述通式(i)的化合物。
[0220]
[表3]
[0221][0222]
*1:原料是指上述通式(i)的化合物。
[0223]
根据上述表2和表3可以理解,根据本发明的方法,能够由式(i)的化合物以提高的收率得到高纯度的式(iii)的化合物。
[0224]
产业上利用的可能性
[0225]
根据本发明的制造方法,能够以提高的收率制造高纯度的式(iii)的化合物。此外,本发明的制造方法在麝香烯酮的制造方法中是有用的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。