核糖体图谱分析中rna片段的去除方法及应用
技术领域
1.本发明涉及核糖体图谱分析领域,具体而言,涉及一种核糖体图谱分析中rna片段的去除方法及应用。
背景技术:
2.经典的中心法则展示了基因从dna转录成rna,rna翻译成蛋白,最终通过蛋白行使功能的过程。蛋白表达水平是基因功能的重要体现之一,目前检测蛋白表达可以通过elisa、质谱和核糖体图谱分析进行评估。核糖体图谱分析(ribosome profiling,ribo-seq)促进了多样化且复杂的生物过程中基因表达调控层面的新发现,是蛋白质合成机制,乃至新蛋白质研究的重要手段之一,为编码区域的实验注释提供了一个系统化的方法。
3.核糖体图谱分析是通过rna酶处理细胞裂解物,把不受核糖体保护的rna降解掉,随后分离被核糖体保护的mrna片段。通过这一实验处理可得到30个核苷酸左右的rna序列,可将其直接定位(map)到原始的mrna,用于确定翻译中的核糖体的准确位置。核糖体图谱用于构建链特异性文库并进行高通量测序,再将这些测序片段map到合适的基因组上。通过对蛋白合成的比率和mrna的丰度比较,可检测每个mrna的翻译效率。
4.但在核糖体图谱数据中,包括人样本的核糖体图谱数据中,除了rrna残留外还有大量的trna片段,这些trna片段数据平均占据整体过滤后数据(clean reads)的30%以上,且现有技术没有能够去除相关的trna的方法和试剂。
技术实现要素:
5.本发明的主要目的在于提供一种核糖体图谱分析中rna片段的去除方法及应用,以解决现有技术中难以去除ribo-seq文库中的trna片段的问题。
6.为了实现上述目的,根据本发明的第一个方面,提供了一种trna去除探针,该trna去除探针包括能够与seq id no:1所示的保守序列特异性结合的序列。
7.进一步地,trna去除探针包括由脱氧核糖核苷酸组成的dna序列。
8.进一步地,trna去除探针包括含有seq id no:2所示序列的序列;优选地,trna去除探针的5’端为seq id no:2所示序列;优选地,trna去除探针为seq id no:2所示的序列。
9.进一步地,trna去除探针的核糖核苷酸上具有锁核酸修饰;优选地,锁核酸修饰的个数为1-10个,更优选为3-8个,进一步优选为5-7个;优选地,trna去除探针的长度为18-29nt,更优选为19-25nt,进一步优选为22-24nt。
10.为了实现上述目的,根据本发明的第二个方面,提供了一种核糖体图谱分析中rna片段的去除方法,该去除方法包括:利用上述trna去除探针与核糖体图谱分析样本结合,trna去除探针与rna片段中的trna片段形成双链rna结构,通过去除双链rna结构实现对于rna片段的去除。
11.进一步地,去除双链rna结构的方法包括利用磁珠、rna酶、层析柱或过滤膜中的一种或多种去除双链rna结构。
12.进一步地,去除方法包括:在核糖体图谱分析文库的建库过程中,将trna去除探针与连接有测序接头的核糖体图谱分析样本结合;核糖体图谱分析文库的建库过程包括:将细胞或组织样本进行裂解,利用翻译抑制剂固定核糖体,获得核糖体固定样本;利用rna酶消化核糖体固定样本,纯化获得核糖体保护片段;将核糖体保护片段进行末端磷酸化和测序接头连接,获得连接有测序接头的核糖体图谱分析样本;将trna去除探针与连接有测序接头的核糖体图谱分析样本结合,去除双链rna结构,获得去除trna样本;将去除trna样本进行反转录、pcr富集和纯化,获得去除trna的核糖体图谱分析文库。
13.进一步地,去除方法还包括利用用于去除rrna的探针,去除rna片段中的rrna片段;优选地,trna去除探针与用于去除rrna的探针同时与核糖体图谱分析样本结合,trna去除探针与trna片段形成第一双链rna结构,用于去除rrna的探针与rrna片段形成第二双链rna结构,同时去除第一双链rna结构和第二双链rna结构。
14.为了实现上述目的,根据本发明的第三个方面,提供了一种rna去除试剂盒,该rna去除试剂盒包括上述trna去除探针。
15.进一步地,rna去除试剂盒还包括用于去除rrna的探针。
16.为了实现上述目的,根据本发明的第四个方面,提供了一种上述trna去除探针、去除方法或rna去除试剂盒在核糖体图谱分析或核糖体保护trna分析中的应用。
17.应用本发明的技术方案,利用上述能够与seq id no:1所示的保守序列特异性结合的trna去除探针,能够去除核糖体图谱分析中被核糖体保护的trna片段,提高ribo-seq文库的有效数据的比例。
附图说明
18.构成本技术的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
19.图1示出了根据本发明实施例3的核糖体图谱分析文库的建库过程示意图。
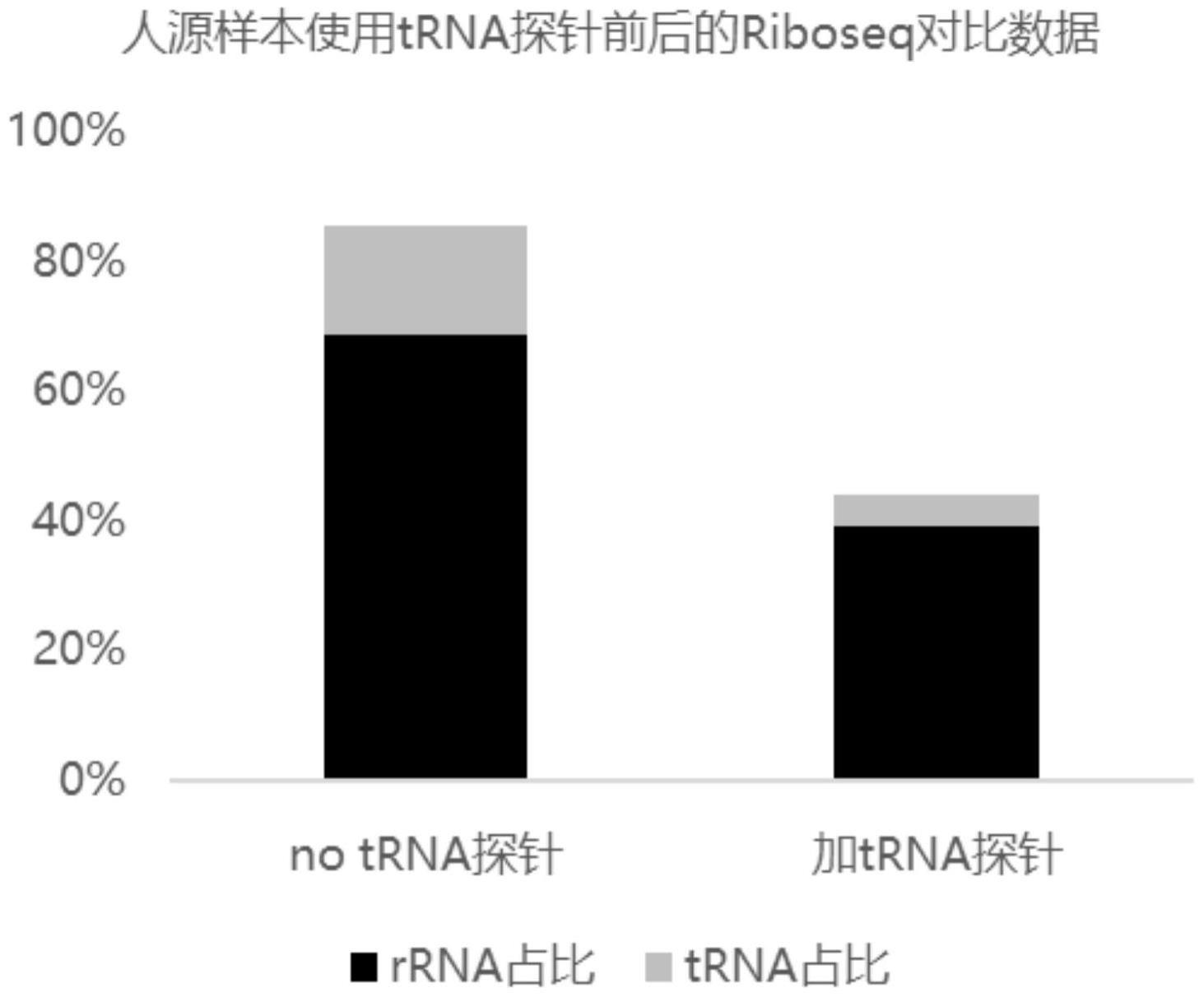
20.图2示出了根据本发明实施例6的对于24个人源样本处理前后的结果图。
具体实施方式
21.需要说明的是,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
22.如背景技术所提到的,在现有的人样本的核糖体图谱数据中,含有大量的trna片段,这些trna片段数据平均占据clean reads的30%以上,占用测序资源,造成测序资源的浪费和成本的提高,并对后续数据分析产生影响。因而,在本技术中发明人尝试开发一种能够去除核糖体图谱分析中trna片段的trna去除探针,基于该探针提出了本技术的一系列保护方案。
23.在本技术第一种典型的实施方式中,提供了一种trna去除探针,该trna去除探针包括能够与seq id no:1所示的保守序列特异性结合的序列。
24.seq id no:1:ucccugguggucuagugguuaggauucgg。
25.上述seq id no:1所示的序列为人样本中的人源核糖体保护的trna片段保守序列。上述保守序列为核糖核苷酸组成的rna序列。利用能够与seq id no:1所示的保守序列
特异性结合的探针,此种探针能够与核糖体图谱分析中trna片段(包括但不限于保守序列)特异性结合形成双链结构,通过去除双链结构能够实现对于样本中trna片段的去除。探针包括但不限于由核糖核苷酸组成的rna序列、由脱氧核糖核苷酸组成的dna序列或由脱氧核糖核苷酸和核糖核苷酸共同组成的dna-rna序列。
26.上述seq id no:1所示的保守序列与人的转录本数据库进行比对,仅匹配到一条lncrna。即上述序列不能匹配到基因编码区,能够与上述保守序列互补配对的序列不会对含有编码信息的mrna产生影响,不影响核糖体图谱分析的准确性。
27.上述trna去除探针能够与seq id no:1所示的保守序列的全部或部分序列进行特异性结合,“部分特异性结合”包括与保守序列中的一部分进行特异性结合,“一部分”包括连续或不连续的10个或更多个碱基,包括但不限于与保守序列上的10、12、14、15、16、17、18、19、20、21、22、23、24、25、26、27或28个碱基特异性结合。
28.在一种优选的实施例中,trna去除探针包括由核糖核苷酸组成的rna序列。
29.在一种优选的实施例中,trna去除探针包括含有seq id no:2所示序列的序列;优选地,trna去除探针的5’端为seq id no:2所示序列;优选地,trna去除探针为seq id no:2所示的序列。
30.seq id no:2:ccgaatcctaaccactagaccac。
31.上述seq id no:2为脱氧核糖核苷酸组成的dna序列。seq id no:2能够与上述seq id no:1特异性结合,形成双链结构。trna去除探针中含有seq id no:2所示的序列。优选地,在trna去除探针中,seq id no:2位于该trna去除探针的5’端,在seq id no:2的3’端可选地设置有其他能够与保守序列互补配对的核糖核苷酸。
32.在一种优选的实施例中,trna去除探针的核糖核苷酸上具有锁核酸修饰;优选地,锁核酸修饰的个数为1-10个,更优选为3-8个,进一步优选为5-7个;优选地,锁核酸修饰的核糖核苷酸位于trna去除探针5’端第1位至第23位中的任意一个或多个位置,包括但不限于trna去除探针5’端第1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22或23位;优选地,锁核酸修饰的核糖核苷酸位于trna去除探针5’端第3位、6位、9位、12位、15位或19位中的任意一个或多个位置;优选地,trna去除探针的长度为18-29nt,包括但不限于18、19、20、21、22、23、24、25、26、27、28或29nt,更优选为19-29nt,19-25nt,22-29nt,进一步优选为22-24nt。
33.上述trna去除探针中的核糖核苷酸可选地为具有锁核酸(lna,locked nucleic acid)结构的核糖核苷酸,上述锁核酸能够提高trna去除探针与样本中trna片段结合的特异性,防止错配导致的核糖体图谱分析中编码信息的丢失。
34.在本技术第二种典型的实施方式中,提供了一种核糖体图谱分析中rna片段的去除方法,该去除方法包括:利用上述trna去除探针与核糖体图谱分析样本结合,trna去除探针与rna片段中的trna片段形成双链rna结构,通过去除双链rna结构实现对于trna片段的去除。
35.在一种优选的实施例中,去除双链rna结构的方法包括利用磁珠、rna酶、层析柱或过滤膜中的一种或多种去除双链rna结构。
36.在一种优选的实施例中,去除方法包括:在核糖体图谱分析文库的建库过程中,将trna去除探针与连接有测序接头的核糖体图谱分析样本结合;核糖体图谱分析文库的建库
reads的种类和丰度,将按照丰度排名后的trna序列保留下来,随后对筛选出来的trnareads序列进行命名,样本编号 reads占比作为序列名称进行区分,然后将序列转换成fasta格式,使用mafft软件进行多重序列相似性比对,将比对完的结果文件导入bioedit或其他序列编辑软件,对序列文件进行分类,得到四个排序靠前序列:
47.seq id no:1:ucccugguggucuagugguuaggauucgg;
48.seq id no:5:gcauuggugguucagugguagaauucucgc;
49.seq id no:6:ucccauauggucuagcgguuaggauuccugg;
50.seq id no:7:gccgugaucguauagugguuaguacu。
51.seq id no:1、seq id no:5、seq id no:6和seq id no:7在trna数据中的占比分别是75%、12%、7%和5%。考虑到后续针对这4条trna序列的探针设计是否对rpf(核糖体保护片段)的有影响,设计过程中也对上述4条trna序列进行mrna比对,发现仅可以比对到一条lncrna序列,对rpf无影响,因此对丰度占比较高的seq no1序列进行探针设计并应用在后续的riboseq建库过程中。
52.实施例2 ribo-seq中trna去除探针设计
53.1.1样本信息和trna reads丰度分析
54.选择20个人细胞样本的ribo-seq测序数据进行分析,分析每个样本的trnareads的种类和丰度,将丰度排名top10的trna序列保留下来(top10的reads数占总reads数的60%以上),做下一步分析。
55.1.2 trna reads相似性分析
56.对上一步筛选出来的trna reads序列进行命名,样本编号 reads占比作为序列名称进行区分,然后将序列转换成fasta格式,使用mafft软件进行多重序列相似性比对。
57.将比对完的结果文件导入bioedit或其他序列编辑软件,对序列文件进行分类,分类原则:差异位点≤1个归为一类,筛选出如下序列:
58.seq id no:1:ucccugguggucuagugguuaggauucgg,占比在70%以上,作为人样本trna去除的保守序列。
59.1.3序列特异性分析
60.采用bowtie将seq id no:1序列与人的转录本数据库进行比对,匹配到一条lncrna。上述序列未匹配到基因编码区,不会对mrna序列产生影响。
61.1.4trna去除探针序列设计
62.针对上述seq id no:1,设计能够与seq id no:1互补配对的seq id no:2探针。
63.seq id no:2:ccgaatcctaaccactagaccac。
64.seq id no:2的信息如表1所示。
65.表1:trna去除探针序列信息
66.名称序列长度纯化方式od260质量(μg)nmolgc(%)tm(℃)mwhsaseq id no:223nthplc132.64.652.257.17116.6
67.其中,seq id no:2的第3、6、9、12、15和19位的核糖核苷酸上具有锁核酸修饰;
68.seq id no:2为由脱氧核糖核苷酸组成的dna序列。
69.实施例3
70.1.样本裂解
71.1.1组织样本裂解
72.1)准备研磨珠、2.0ml研磨管,将样本切碎放入研磨管中并放入液氮中速冻。
73.2)使用自动研磨器,在48hz,20s的条件下研磨2次,将组织研磨成粉末状。
74.3)试剂准备,配方如表2和表3所示。
75.表2多聚体缓冲液(polysome buffer)配方
76.试剂使用量(μl)/样本储存条件(℃)1m tris-cl ph7.520常温5m nacl3041m mgcl25常温nf-h2o9454总体积1000 77.表3动物组织和细胞裂解液配方
78.试剂使用量(μl)/样本储存条件(℃)polysome buffer878410%triton x-100100-20100mm dtt10-20dnasel(1u/μl)10-2050mg/ml放线菌酮(cycloheximide)2-20总体积1000 79.4)加入1ml裂解液,枪头充分吹打混匀。
80.5)冰上裂解20min,期间每2min轻微震荡混匀。
81.6)4℃,13000rpm离心10min,将上清转移至冰上预冷的新离心管中。
82.7)qubit测量裂解液浓度并记录,初浓度≥100ng/μl即可进行下步反应。
83.1.2细胞样本裂解
84.1)明确细胞样本类型,若为冻存液保存的细胞,需在37℃水浴复苏细胞后常温500g 5min离心收集细胞,弃去保护液后进行裂解。
85.2)加入600μl细胞裂解液,枪头充分吹打混匀。
86.3)5号注射器反复吹打细胞裂解液8次。
87.4)冰上裂解20min,期间每2min轻微震荡混匀。
88.5)4℃,13000rpm离心10min,将上清转移至冰上预冷的新离心管中。
89.6)qubit测量裂解液浓度并记录,初浓度≥100ng/μl即可进行下步反应。
90.2.核糖体保护片段(rpf)富集
91.2.1试剂和耗材准备
92.所用试剂如表4所示。
93.表4
[0094][0095]
2.2rna酶消化
[0096]
1)取上步裂解液200μl,依据rna总量计算需要的rnasei,加入7.5μl rnasei(100u/μl)。
[0097]
注:裂解液浓度在100-200ng/μl时,200μl裂解液加7.5μl rnase i。
[0098]
2)不同样本类型酶切环境不一样,4℃混匀仪孵育1h。
[0099]
3)孵育30min后,可准备microspins-400 columns树脂柱。
[0100]
2.3rna酶消化终止
[0101]
加入10μl superase*in rnase inhibitor,充分混匀,终止核酸酶消化反应。
[0102]
2.4microspin s-400column过滤
[0103]
1)每个样本准备3ml polysome buffer。
[0104]
2)将microspin s-400column颠倒几次混匀树脂,轻弹色谱柱以除去树脂中的气泡。
[0105]
3)打开色谱柱上下两端,加入缓冲液(polysome buffer),使缓冲液在重力作用下滴出。
[0106]
4)连接收集管,在固定角度的台式离心机中,600g室温离心4min。弃去流出的缓冲液并将柱子转移到新的2mlep管中。
[0107]
5)立即取200μl核酸酶消化后的rpf样本(确认已添加rnase inhibitor)在600g离心2min。收集流出液。
[0108]
6)加20μl的10%sds(每100μl加入10μl的10%sds),用作rpf rna后续处理。
[0109]
7)若上述混合物大于220μl,则平分成两管,并按照2.5的试剂配比配置体系。
[0110]
2.5 rna clean&concentrator-25kit纯化rpf rna样本
[0111]
1)使用试剂盒(zymo research,cat#r1017)进行试验,使用前确定rnawash buffer中已加入乙醇。
[0112]
2)按下表5配制体系并混匀。
[0113]
表5 rna clean&concentrator-25kit纯化体系
[0114]
试剂使用量(μl)/样本rna binding buffer220无水乙醇495rpf样本110
[0115]
3)将zymo-spin
tm iic column放置在收集管中,将上述体系转移至柱子中,室温13000rpm离心1min,弃收集液。
[0116]
4)重复上述操作,将剩余体系转移至柱子中,室温13000rpm离心1min,弃收集液。
[0117]
5)加入400μl rnaprep buffer到柱子中,室温13000rpm离心1min,弃收集液。
[0118]
6)加入700μl rnawash buffer到柱子中,室温13000rpm离心1min,弃收集液。
[0119]
7)加入400μlrnawash buffer到柱子中,室温13000rpm离心2min,弃收集液,将柱子转移至新的无rna酶的1.5ml离心管中。
[0120]
8)将18μlnf-h2o小心加入回收柱中央,室温静置2min,室温13000rpm离心2min。
[0121]
9)用qubit定量并记录总的rna量。
[0122]
3.变性page切胶
[0123]
3.1试剂准备
[0124]
所用试剂如表6所示。
[0125]
表6
[0126]
试剂储存条件(℃)尿素常温30%聚丙烯酰胺410
×
tbe4dd h2o常温10%过硫酸铵4temed4denaturing gel loading dye(thermofisher,r0641)-2020/100oligo ladder(1ng/μl)(thermofisher,10597012)-20sybr gold(thermofisher,s11494)常温
[0127]
3.2配制15%变性page胶
[0128]
15%变性page胶配方如表7所示。
[0129]
表7
[0130]
试剂15ml胶用量30ml胶用量尿素6.3g12.6g30%聚丙烯酰胺6ml12mli0
×
tbe1.5ml3mldd h2o7.5ml15ml10%过硫酸铵75μl150μltemed15μl30μl
[0131]
3.3准备样本和marker(20/100oligo ladder)
[0132]
1)10μl样本与10μl2x loading dye(denaturing gel loading dye)混匀,总体积20μl。
[0133]
2)8μl20/100oligo ladder与10μl 2x loading dye混匀,5μl28/30nt marker与5μl 2xloading dye混匀。
[0134]
28/30nt marker由2条rna序列组成,其中:
[0135]
28nt序列为5
’‑
auguacacggagucgacccgcaacgcga-3’(seq id no:3),
[0136]
30nt序列为5
’‑
auguacacggagucgaaacccgcaacgcga-3’(seq id no:4),
[0137]
28nt序列和30nt序列的3’端均具有磷酸化修饰(phos)。
[0138]
3)pcr仪75℃,3min。使样本和20/100 oligo ladder变性,结束后立即置于冰上。
[0139]
3.4电泳和染色
[0140]
180v电泳50min~1h,电泳完成后,将胶切角标记后取下,使用sybr gold染色5min,1∶6000(30ml纯水 5μl sybr gold)。暗室蓝光观察rna条带。
[0141]
3.5切胶
[0142]
切胶,范围28-30nt。将切取的胶转移至扎好孔的离心管中。
[0143]
4.核糖体保护片段(rpf)回收纯化
[0144]
4.1耗材准备
[0145]
取0.5ml离心管,用注射器针头在底部扎孔呈筛状,置于2ml离心管中。准备spin-x过滤管。
[0146]
4.2试剂准备
[0147]
所用试剂如表8所示。
[0148]
表8
[0149]
试剂储存条件(℃)nf h2o常温3m醋酸钠常温糖原(glycogen)-20无水乙醇-2080%乙醇-20,现配现用
[0150]
4.3回收纯化
[0151]
1)将切下来的胶放入准备好的2ml离心管中(套0.5ml离心管),4℃13000rpm离心5min。
[0152]
2)向凝胶碎末中加入200μlnf-h2o,震荡器37℃,1000rpm摇1h。
[0153]
3)将上述溶液转移至spin-x离心过滤管中,4℃,13000rpm离心10min。
[0154]
4)按下表9加入试剂并充分混匀。
[0155]
表9
[0156]
试剂使用量(μl)/样本3m醋酸钠13glycogen2.5无水乙醇600
[0157]
5)瞬时离心,置于-20℃放置1h以上。
[0158]
6)4℃13000rpm离心15min,小心吸取上清。
[0159]
7)加入1000μl70%乙醇,13000rpm离心5min后弃上清。
[0160]
8)重复第7步,尽可能吸走上清。
[0161]
9)用43μl nf-h2o溶解rna沉淀,轻柔吹吸20次促进溶解。
[0162]
5.磷酸化(末端修复)
[0163]
5.1试剂准备
[0164]
所用试剂如表10所示。
[0165]
表10
[0166]
试剂储存条件(℃)t4 pnk buffer(neb,m0201v)-20superase*in rnase inhibitor(20u/μl)(thermofisher,am2694)-20t4 pnk(10u/μl)(neb,m0201v).20无水乙醇4nf h2o常温
[0167]
5.2末端磷酸化
[0168]
1)按下表11依次添加试剂:
[0169]
表11末端修复体系
[0170] 体积(μl)rna样本43t4 pnk buffer5superase*in rnase inhibitor(20u/μl)1t4 pnk(10u/μl)1
[0171]
2)震荡混匀,瞬时离心,pcr仪上反应:37℃,1h;70℃,10min。
[0172]
3)向样本中加入nf-h2o,将样本体积补足至100μl,按下表12配制体系并混匀。
[0173]
表12
[0174]
试剂使用量(μl)/样本3m醋酸钠13glycogen2.5无水乙醇300
[0175]
4)瞬时离心,置于-20℃放置1h以上。
[0176]
5)4℃13000rpm离心15min,小心吸取上清。
[0177]
6)加入500μl70%乙醇,13000rpm离心5min后弃上清。
[0178]
8)重复第7步,尽可能吸走上清。
[0179]
9)待沉淀晾干2min后,用7μlnf-h2o溶解样本。
[0180]
6.接头连接
[0181]
6.1试剂准备
[0182]
所用试剂如表13所示,本技术建库部分涉及的试剂来源于neb试剂盒,货号为neb#e7300s。
[0183]
表13
[0184]
试剂储存条件(℃)3
′
sr adaptor for illumina(稀释4倍)-20nf h2o常温3
′
ligation reaction buffer(2x)-203′
ligation enzyme mix-20sr rt primer for illumina(稀释一倍)-205
′
sr adaptor for illumina(denatured)-205
′
ligation reaction buffer(10x)-205
′
ligation enzyme mix-20
[0185]
6.23’接头连接
[0186]
1)将末端修复后的样本放入新的pcr管中,加入下列表14所示的反应体系,充分混匀。
[0187]
表14
[0188][0189]
2)pcr仪70℃变性2min之后立即放冰上。
[0190]
3)向上述体系中加入下列表15所示的反应液
[0191]
表15
[0192]
试剂使用量(μl)3
′
ligation reaction buffer(2x)53
′
ligation enzyme mix1.5total volume14
[0193]
4)25℃孵育1h,之后立即放冰上。
[0194]
6.3反转录引物杂交(除游离3’接头)
[0195]
1)将以下表16所示组分添加到连接混合物中并充分混合。
[0196]
表16
[0197]
试剂使用量(μl)sr rt primer for illumina(稀释一倍)1.5total volume15.5
[0198]
2)反应条件如表17所示。
[0199]
表17
[0200]
温度时间75℃5min37℃15min25℃15min
4℃hold
[0201]
6.45’接头连接
[0202]
1)取1μl5
′
sr adaptor(存放在-80℃)至新的pcr管中,70℃变性2min,之后立刻放置在冰上。
[0203]
注意:第一次使用5
′
sr adaptor先离心,再加入120μl的depc水溶解,分装至pcr管中,每管10μl,放于-80℃储存。
[0204]
1)向上管反应体系中加入下列表18所示反应溶液,充分混匀;
[0205]
表18
[0206]
试剂使用量(μl)5
′
sr adaptor for illumina(denatured)0.55
′
ligation reaction buffer(10x)0.55
′
ligation enzyme mix1.25total volume17.25
[0207]
4)25℃孵育1小时,之后立刻放在冰上。
[0208]
7.去除rrna和trna
[0209]
7.1去除rrna
[0210]
在上一步样本中添加1μlqiaseqfastselect-rrnahmr和1μltrna去除探针(1ng),充分混匀,在热循环仪中执行以下表19所示程序:
[0211]
表19
[0212]
阶段温度(℃)时间(min)175227023652460255526372725284hold
[0213]
7.2磁珠纯化
[0214]
1)准备xp磁珠,提前取出,室温放置30min,使用之前旋涡振荡充分混匀,吸取110μl(2.2
×
)加入到50μl下机的样本中,用移液器吹打10次充分混匀,室温静置孵育5min;
[0215]
2)将样品置于磁力架上,静置5min,待溶液澄清后,小心移除上清;
[0216]
3)保持样品始终处于磁力架中,加入200μl新鲜配制的80%乙醇漂洗磁珠(注意使用新鲜配制的80%乙醇漂洗磁珠,且不要吹散磁珠),室温孵育30s,小心移除上清;
[0217]
4)重复上一步,总计漂洗磁珠2次;
[0218]
5)保持样品始终处于磁力架中,开盖空气干燥磁珠5min;应避免磁珠过分干燥(龟裂)而降低回收效率;
[0219]
6)将样品从磁力架中取出,加入18μlnf-h2o,轻轻吹打10次充分混匀液体,室温静置5min。将样品置于磁力架上,静置5min(待溶液澄清)后,小心吸取18μl上清至一个新的
nuclease-free pcr管中。
[0220]
8.反转录
[0221]
8.1试剂准备
[0222]
所用试剂如表20所示,所用试剂来源于试剂盒(neb,#e7300s)。
[0223]
表20
[0224]
试剂储存条件(℃)first strand synthesis reaction buffer-20(红盖)murine rnase inhibitor-20(红盖)protoscriptllreverse transcriptase-20(红盖)
[0225]
8.2反转录
[0226]
1)向5’接头连接产物中加入以下表21所示成分。
[0227]
表21
[0228]
试剂使用量(μl)first strand synthesis reaction buffer4murine rnase inhibitor0.5protoscriptllreverse transcriptase0.5total volume22.25
[0229]
2)在50℃下孵育1小时,立即进行pcr扩增。
[0230]
9.pcr富集
[0231]
9.1试剂准备
[0232]
所用试剂如表22所示。
[0233]
表22
[0234]
试剂储存条件(℃)longamp taq 2xmaster mix(neb,#e7300s)-20sr primer for illumina(neb,#e7300s)-20index(x)primer(neb,#e7300s)-20nuclease-free water常温
[0235]
9.2pcr富集
[0236]
1)将以下表23所示组分添加到rt反应混合物中并混匀。
[0237]
表23
[0238]
试剂使用量(μl)longamp taq 2xmaster mix25sr primer for illumina1.5index(x)primer1.5nuclease-free water2.5total volume52.75
[0239]
2)pcr循环条件如表24所示。
[0240]
表24
[0241][0242]
3)qubit定量并记录。
[0243]
10.非变性page切胶
[0244]
1)配制12%的非变性page,比例如表25所示。
[0245]
表25
[0246]
试剂15ml胶用量30ml胶用量30%聚丙烯酰胺6 ml12ml10xtbe1.5 ml3mlddh2o7.5ml15ml10%过硫酸铵75μl150μltemed15μl30μl
[0247]
2)取50μlpcr产物加入10μl 6x loading buffer(neb,#e7300s),一板胶准备5μl20bp-500bp的maker。
[0248]
3)按需求对样本进行编号,依次点样。
[0249]
4)150v电泳100min。
[0250]
5)电泳完成后,将胶切角标记后取下,使用sybr gold染色5min,1∶6000(30ml纯水 5μl sybr gold)。
[0251]
6)切胶,范围140-160bp。将切取的胶转移至扎好孔的离心管中。
[0252]
11.建库产物回收纯化
[0253]
11.1耗材准备
[0254]
取0.5ml离心管,用注射器针头在底部扎孔呈筛状,置于2ml离心管中。准备spin-x过滤管。
[0255]
11.2试剂准备
[0256]
所用试剂如表26所示。
[0257]
表26
[0258]
试剂储存条件(℃)nf h2o常温3m醋酸钠常温
glycogen-20无水乙醇-2080%乙醇-20,现配现用
[0259]
11.3回收纯化
[0260]
1)将切下来的胶放入准备好的2ml离心管中(套0.5ml离心管),4℃13000rpm离心5min。
[0261]
2)向凝胶碎末中加入200μleb(gel elution buffer),震荡器50℃1000rpm摇1h。
[0262]
3)将上述溶液转移至spin-x离心过滤管中,4℃13000rpm离心10min。
[0263]
4)按下表27加入试剂并充分混匀。
[0264]
表27
[0265]
试剂使用量(μl)/样本3m醋酸钠13glycogen2.5无水乙醇600
[0266]
5)瞬时离心,置于-20℃放置1h以上。
[0267]
6)4℃13000rpm离心15min,小心吸取上清。
[0268]
7)加入1000μl 70%乙醇,13000rpm离心5min后弃上清。
[0269]
8)重复第7步,尽可能吸走上清。
[0270]
9)用11μl eb溶解dna沉淀,轻柔吹吸20次促进溶解。
[0271]
10)qubit定量并记录,在每个文库管盖上写上文库名称,将文库稀释至1ng/μl,取20μl稀释液送库检。
[0272]
上述核糖体图谱分析文库的建库过程示意图如图1所示。
[0273]
实施例4
[0274]
选择人细胞样本,利用实施例3所示的方法进行测试,结果如表28所示,结果显示数据中trna片段的残留效果显著被清除掉,降低了无效数据的产出,人细胞样本trna片段残留维持在5%以下。
[0275]
表28:人细胞样本trna去除探针序列有效性测试结果
[0276][0277]
注:在表28中,对于t1-1和t1-2样本中trna片段的残留的检查结果,虽然t1-1样本中加入的去除探针的量更多,但t1-1样本中trna的残留量大于t1-2样本。此种现象属于在实验中可能存在的正常实验现象。对于样本中的trna去除的效率是一个范围,上述trna去除探针和去除方法的使用,能够大幅降低样本中的trna含量。在对trna去除的过程中可能与实验操作、反应体系添加试剂等细微因素有关,对于单纯的去除后残留量数据之间的比较实际意义不大。
[0278]
实施例5
[0279]
利用实施例3所示的方法对人相关细胞和组织进行测试,结果显示trna探针的去除效果表现超出预期,有效性上非常高,可以使trna的片段残留维持在10%以内,具体实验结果如表29所示。
[0280]
表29
[0281]
样本类型rrna占比trna占比rrna trna占比coding%人源胚胎干细胞34.79%3.34%38.13%89.60%人源胚胎干细胞37.36%3.86%41.22%86.92%人甲状腺癌乳头状细胞37.64%2.76%40.40%87.23%人甲状腺癌乳头状细胞38.79%2.58%41.37%87.61%人胃癌组织40.65%5.28%45.93%83.76%人胃癌组织45.47%2.32%47.79%88.74%
[0282]
实施例6
[0283]
利用上述实施例3所示的方法对24个人源样本进行测试,使用24个人源样本进行riboseq实验,对比加入或不加入trna探针,汇总后的结果如图2所示。综合结果显示加入trna探针可以有效降低rrna和trna残留。对于rrna残留的降低,申请人推测是上述trna探针还能够与部分残留的rrna结合,从而实现在去除trna的同时也去除部分rrna的预料不到的技术效果。
[0284]
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:利用上述能够与seq id no:1所示的保守序列特异性结合的trna去除探针,包括但不限于seq id no:2所示的trna去除探针,或利用上述去除方法或试剂盒,均能够去除核糖体图谱分析中被核糖体保护的trna片段,提高ribo-seq文库的有效数据的比例。
[0285]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。