1.本发明涉及纤维增强复合材料用的环氧树脂组合物、使用了该环氧树脂组合物的纤维增强复合材料、以及纤维增强复合材料的制造方法。
背景技术:
2.由增强纤维和基体树脂构成的纤维增强复合材料由于可以进行发挥了增强纤维和基体树脂的优点的材料设计,因此以航空宇宙领域为代表,用途扩大到体育领域、一般产业领域等。
3.作为增强纤维,可使用玻璃纤维、芳族聚酰胺纤维、碳纤维、硼纤维等。作为基体树脂,热固性树脂、热塑性树脂都可使用,但往往使用向增强纤维的含浸容易的热固性树脂。作为热固性树脂,可使用环氧树脂、不饱和聚酯树脂、乙烯基酯树脂、酚醛树脂、双马来酰亚胺树脂、氰酸酯树脂等。
4.作为纤维增强复合材料的成型方法,可应用预浸料法、手糊法、纤维缠绕法、拉挤成型法、rtm(树脂传递模塑,resin transfer molding)法等方法。预浸料法是通过将在增强纤维中含浸了环氧树脂组合物的预浸料叠层为所希望的形状,进行加热从而获得成型物的方法。该预浸料法虽然面向具有在航空器、汽车等结构材料用途中被要求的高材料强度的纤维增强复合材料的生产,但需要经过预浸料的制作、叠层等多个工艺,因此只能少量生产,不适合于大量生产,生产性存在问题。另一方面,rtm法是在配置在加热了的成型模内的增强纤维基材中注入液态的环氧树脂组合物,使其含浸,在该成型模内加热固化而获得成型物的方法。如果是该方法则通过准备成型模从而不仅可以不经由预浸料制作工序而在短时间将纤维增强复合材料成型,而且也具有即使是复杂形状的纤维增强复合材料也能够容易地成型这样的优点。
5.作为液态的环氧树脂组合物,可使用单组分型或双组分型环氧树脂组合物。所谓单组分型环氧树脂组合物,是包含环氧树脂、固化剂,全部成分被预先混合成1个的环氧树脂组合物。与此相对,将由包含环氧树脂作为主成分的环氧主剂液和包含固化剂作为主成分的固化剂液构成,在即将使用前将环氧主剂液与固化剂液的2液混合而获得的环氧树脂组合物称为双组分型环氧树脂组合物。
6.在rtm法中,往往使用单组分型环氧树脂组合物。为了将单组分型环氧树脂组合物应用于航空器主翼、尾翼等巨大的结构材料,要求同时满足接下来举出的2个条件的物质。第一,为了获得没有未含浸的巨大的结构材料,要求向增强纤维基材的树脂注入中的树脂粘度长时间保持低粘度,第二,对于巨大的结构材料,有时加热固化时的升温速度慢,因此要求即使在该情况下也能够将在结构材料用途中被要求的高水平的物性(耐热性、高压缩强度、耐冲击性、耐久性)赋予纤维增强复合材料。
7.对于这样的现状,公开了包含亚甲基双(3-氯-2,6-二乙基苯胺)(m-cdea)作为固化剂的单组分型环氧树脂组合物,提出了抑制长时间粘度的上升的方法(专利文献1)。进一
步,公开了芴胺固化剂作为一部分固体而分散了的单组分型环氧树脂组合物,提出了抑制长时间粘度的上升的方法(专利文献2)。此外,公开了包含4,4
’‑
亚甲基双(2-异丙基-6-甲基苯胺)(m-mipa)、4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺)(m-dipa)的单组分型环氧树脂组合物,提出了在180℃下可以进行充分的高速固化,并且在成型后的脱模工序时,树脂充分固化,可以赋予高水平的纤维增强复合材料物性的方法(专利文献3、4)。
8.现有技术文献
9.专利文献
10.专利文献1:日本专利第5808057号公报
11.专利文献2:日本特表平11-511503号公报
12.专利文献3:日本特开2010-150310号公报
13.专利文献4:国际公报第2020/008847号
技术实现要素:
14.发明所要解决的课题
15.对于上述专利文献1所记载的方法,虽然能够抑制长时间粘度的上升,但是由于仅使用反应性低的亚甲基双(3-氯-2,6-二乙基苯胺)(m-cdea),因此具有下述课题:固化性低,在加热固化时的升温速度慢的情况下,有时将增强纤维基材连结的粘合剂在树脂中过度熔融,纤维增强复合材料的层间厚度变得不均匀,不表现充分的冲击后压缩强度。
16.对于上述专利文献2所记载的方法,虽然能够抑制长时间粘度的上升,但是具有下述课题:有时作为一部分固体而分散的芴胺固化剂凝集从而有时在向增强纤维基材注入树脂时该凝集物被基材滤出;芴胺固化剂的熔点为201℃,非常高,即使在180℃的高温下有时也一部分溶解残留,因此发生固化不良,不表现充分的高耐热性。
17.对于上述专利文献3、4所记载的方法,虽然能够进行充分的高速固化,并且成型后的树脂充分固化,可以将高耐热性、高机械特性赋予纤维增强复合材料,但具有下述课题:在加热固化时的升温速度慢的情况下,将增强纤维基材连结的粘合剂的表面在树脂中不能熔融,树脂与粘合剂的粘接性差,关于纤维增强复合材料,不表现充分的压缩强度、冲击后压缩强度、耐微裂纹性。
18.这样,对于现有技术,不存在同时满足上述2个条件的单组分型环氧树脂组合物。因此,本发明的目的是提供在向增强纤维基材的树脂注入中长时间保持低粘度,即使在加热固化时的升温速度慢的情况下也可以将在结构材料用途中被要求的高水平的物性(耐热性、高压缩强度、耐冲击性、耐久性)赋予纤维增强复合材料的环氧树脂组合物。进一步,通过使用这样的环氧树脂组合物,从而提供树脂固化物的玻璃化转变温度、湿热时的0
°
压缩强度、冲击后压缩强度、耐微裂纹性优异的纤维增强复合材料。
19.用于解决课题的手段
20.为了解决上述课题,本发明的纤维增强复合材料用环氧树脂组合物具有以下构成。即,一种纤维增强复合材料用环氧树脂组合物,包含相对于全部环氧树脂成分100质量%为70质量%以上且90质量%以下的[a]四缩水甘油基二氨基二苯基甲烷,相对于全部环氧树脂成分100质量%为10质量%以上且30质量%以下的[b]双酚f型环氧树脂,并且包含[c]4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺)和[d]4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基
苯胺)。
[0021]
此外,本发明的纤维增强复合材料包含本发明的纤维增强复合材料用环氧树脂组合物的固化物、和增强纤维基材。
[0022]
发明的效果
[0023]
根据本发明,能够提供在向增强纤维基材的树脂注入中长时间保持低粘度,即使在加热固化时的升温速度慢的情况下也可以将在结构材料用途中被要求的高水平的物性(耐热性、高压缩强度、耐冲击性、耐久性)赋予纤维增强复合材料的纤维增强复合材料用环氧树脂组合物。
具体实施方式
[0024]
以下,对本发明的优选的实施方式进行说明。
[0025]
首先,对本发明中的纤维增强复合材料用环氧树脂组合物进行说明。
[0026]
本发明的纤维增强复合材料用环氧树脂组合物包含相对于全部环氧树脂成分100质量%为70质量%以上且90质量%以下的[a]四缩水甘油基二氨基二苯基甲烷,相对于全部环氧树脂成分100质量%为10质量%以上且30质量%以下的[b]双酚f型环氧树脂,并且包含[c]4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺)和[d]4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺)。以下,有时将上述各成分分别简称为成分[a]、成分[b]、成分[c]、成分[d]。此外,有时将纤维增强复合材料用环氧树脂组合物简称为环氧树脂组合物。
[0027]
通过包含上述质量份的成分[a]、成分[b],并且包含成分[c]、成分[d]的环氧树脂组合物,从而对于现有技术而言困难的向增强纤维基材的树脂注入中的树脂粘度长时间保持低粘度,即使在加热固化时的升温速度慢的情况下也可以将在结构材料用途中被要求的高水平的物性(耐热性、高压缩强度、耐冲击性、耐久性)赋予纤维增强复合材料。
[0028]
本发明中的成分[a]为四缩水甘油基二氨基二苯基甲烷。成分[a]是为了向环氧树脂固化物和纤维增强复合材料给予高耐热性、机械特性而需要的成分。这里所谓成分[a]的四缩水甘油基二氨基二苯基甲烷,是指n,n,n’,n
’‑
四缩水甘油基二氨基二苯基甲烷、或它们的衍生物或异构体。可以举出例如,n,n,n’,n
’‑
四缩水甘油基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二甲基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二乙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二异丙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二-叔丁基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二甲基-5,5
’‑
二乙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二异丙基-5,5
’‑
二乙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二异丙基-5,5
’‑
二甲基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二-叔丁基-5,5
’‑
二乙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二-叔丁基-5,5
’‑
二甲基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3’,5,5
’‑
四甲基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3’,5,5
’‑
四乙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3’,5,5
’‑
四异丙基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3’,5,5
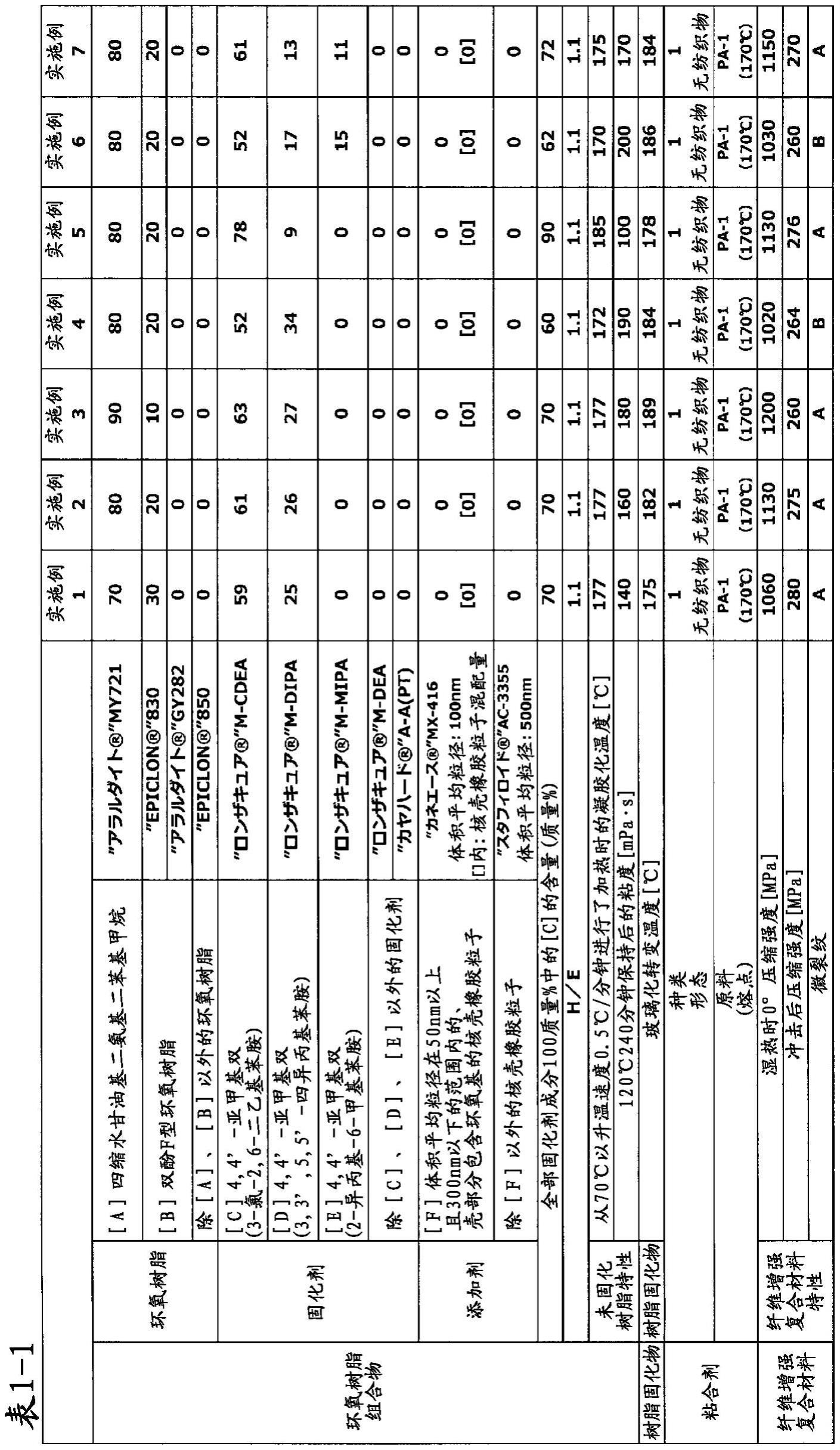
’‑
四-叔丁基-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二氯-4,4
’‑
二氨基二苯基甲烷、n,n,n’,n
’‑
四缩水甘油基-3,3
’‑
二溴-4,4
’‑
二氨基二苯基甲烷等。此外,作为成分[a],可以包含2种以上这些四缩水
甘油基二氨基二苯基甲烷。
[0029]
作为四缩水甘油基二氨基二苯基甲烷的市售品,可举出
“スミエポキシ
(注册商标)”elm434(住友化学工业(株)制)、yh434l(新日铁住金化学(株)制)、“jer(注册商标)”604(三菱化学(株)制)、
“アラルダイト
(注册商标)”my720、
“アラルダイト
(注册商标)”my721(以上,
ハンツマン
·
アドバンズド
·
マテリアルズ
社制)等。
[0030]
本发明中的成分[a]需要在全部环氧树脂成分100质量%中包含70质量%以上且90质量%以下。在全部环氧树脂成分100质量%中包含成分[a]70质量%以上的情况下,环氧树脂固化物表现高耐热性,并且纤维增强复合材料的湿热时的0
°
压缩强度提高。此外,在包含成分[a]90质量%以下的情况下,树脂含浸温度下的环氧树脂组合物的粘度降低,向增强纤维基材的含浸性提高。从这样的观点考虑,成分[a]的含量优选在全部环氧树脂成分100质量%中为80质量%以上且90质量%以下的范围内。需要说明的是,在本发明中,所谓环氧树脂固化物,是指将环氧树脂组合物固化而获得的固化物。
[0031]
本发明中的成分[b]为双酚f型环氧树脂。成分[b]是为了减少树脂含浸温度下的环氧树脂组合物的粘度,使向增强纤维基材的含浸性提高而需要的成分。此外,成分[b]是为了向环氧树脂固化物和纤维增强复合材料给予高机械特性而需要的成分。这里所谓成分[b]的双酚f型环氧树脂,是具有双酚f的2个酚性羟基被缩水甘油基化了的结构的物质。
[0032]
作为双酚f型环氧树脂的市售品,可举出“jer(注册商标)”806、“jer(注册商标)”807、“jer(注册商标)”1750、“jer(注册商标)”4004p、“jer(注册商标)”4007p、“jer(注册商标)”4009p(以上三菱化学(株)制)、“epiclon(注册商标)”830(dic(株)制)、
“エポトート
(注册商标)”ydf-170、
“エポトート
(注册商标)”ydf2001、
“エポトート
(注册商标)”ydf2004(以上新日铁住金化学(株))、
“アラルダイト
(注册商标)”gy282(
ハンツマン
·
ジャパン
(株)制)等。
[0033]
在本发明中,优选成分[b]的50℃下的树脂粘度η(mpa
·
s)满足1000≤η≤10000。在η为1000mpa
·
s以上的情况下,树脂注入温度下的粘度不会过低,在向增强纤维基材的注入时由卷入空气而产生的坑引起的未含浸不易发生。此外,如果在树脂注入温度下,环氧树脂组合物的反应性高,则有时在注入过程中粘度增加,含浸性降低,产生未含浸部,或成型花费时间等,成型变得困难,但在η为10000mpa
·
s以下的情况下,树脂注入温度下的粘度充分低,因此向增强纤维基材的含浸性提高,不易产生未含浸。从这样的观点考虑,更优选树脂粘度η(mpa
·
s)满足1000≤η≤8000。作为树脂粘度η(mpa
·
s)满足上述范围的树脂的市售品,可举出例如,“epiclon(注册商标)”830(dic(株)制)、
“アラルダイト
(注册商标)”gy282(
ハンツマン
·
ジャパン
(株)制)。需要说明的是,本发明中的树脂粘度η(mpa
·
s)按照jis z8803(1991)中的“円
すい
-板形回転粘度計
による
粘度測定方法(采用圆锥-板形旋转粘度计的粘度测定方法)”,使用b型粘度计测定。此外,作为作为烷基取代体的四甲基双酚f型环氧树脂的市售品,可举出
“エポトート
(注册商标)”yslv-80xy(新日铁住金化学(株))等。
[0034]
本发明中的成分[b]需要在全部环氧树脂成分100质量%中包含10质量%以上且30质量%以下。在全部环氧树脂成分100质量%中包含成分[b]10质量份以上的情况下,可以降低树脂含浸温度下的环氧树脂组合物的粘度,使向增强纤维基材的含浸性提高,防止未含浸,进一步在环氧树脂固化物中表现高韧性和弹性模量。此外,在成分[b]为30质量%
以下的情况下,表现高耐热性。从这样的观点考虑,成分[b]的含量优选在全部环氧树脂成分100质量%中为10质量%以上且25质量%以下的范围内。
[0035]
此外,本发明的纤维增强复合材料用环氧树脂组合物可以包含除成分[a]、成分[b]以外的环氧树脂,只要在全部环氧树脂成分100质量%中为20质量%以下即可。作为这样的除成分[a]、成分[b]以外的环氧树脂,可举出除成分[b]以外的双酚型环氧树脂、苯酚酚醛清漆型环氧树脂、甲酚酚醛清漆型环氧树脂、间苯二酚型环氧树脂、苯酚芳烷基型环氧树脂、萘酚芳烷基型环氧树脂、二环戊二烯型环氧树脂、具有联苯骨架的环氧树脂、异氰酸酯改性环氧树脂、四苯基乙烷型环氧树脂、三苯基甲烷型环氧树脂、三缩水甘油基胺型环氧树脂等。可以包含1种也可以包含2种以上除成分[a]、成分[b]以外的环氧树脂。
[0036]
作为除成分[a]、成分[b]以外的环氧树脂,更具体而言,可举出双酚a二缩水甘油基醚、四溴双酚a二缩水甘油基醚、双酚ad二缩水甘油基醚、2,2’,6,6
’‑
四甲基-4,4
’‑
联苯酚二缩水甘油基醚、9,9-双(4-羟基苯基)芴的二缩水甘油基醚、三(对羟基苯基)甲烷的三缩水甘油基醚、四(对羟基苯基)乙烷的四缩水甘油基醚、苯酚酚醛清漆缩水甘油基醚、甲酚酚醛清漆缩水甘油基醚、苯酚与二环戊二烯的缩合物的缩水甘油基醚、联苯芳烷基树脂的缩水甘油基醚、异氰脲酸三缩水甘油酯、5-乙基-1,3-二缩水甘油基-5-甲基乙内酰脲、通过双酚a二缩水甘油基醚与甲苯异氰酸酯的加成而获得的唑烷酮型环氧树脂、苯酚芳烷基型环氧树脂、三缩水甘油基氨基苯酚等。其中除成分[b]以外的双酚型环氧树脂由于易于给予环氧树脂固化物的韧性、耐热性的平衡优异的贡献因此优选使用。特别是液态双酚型环氧树脂由于给予向增强纤维的含浸性优异的贡献,因此作为除成分[a]、成分[b]以外的环氧树脂而优选使用。需要说明的是,在本发明中,所谓“液态”,是指25℃下的粘度为1000pa
·
s以下。此外,所谓“固体状”,是指在25℃下不具有流动性,或流动性极其低,具体而言25℃下的粘度大于1000pa
·
s。这里,粘度按照jis z8803(1991)中的“円
すい
-平板形回転粘度計
による
粘度測定方法(采用圆锥-平板形旋转粘度计的粘度测定方法)”,使用安装了标准锥形转子(1
°
34
’×
r24)的e型粘度计(例如,(株)
トキメック
制tve-30h)而测定。
[0037]
这里,所谓除成分[b]以外的双酚型环氧树脂,是除双酚f以外的双酚化合物的2个酚性羟基被缩水甘油基化了的物质。作为除成分[b]以外的双酚型环氧树脂,可举出双酚a型环氧树脂、双酚ad型环氧树脂、双酚s型环氧树脂等,也包含这些双酚型环氧树脂的双酚化合物部分被卤素取代了的物质、被烷基取代了的物质、被氢化了的物质等。此外,作为双酚型环氧树脂,不限于单体,也可以适合使用具有多个重复单元的高分子量体。从环氧树脂固化物的韧性、耐热性的平衡的观点考虑,在含有除成分[b]以外的双酚型环氧树脂的情况下的含量优选在全部环氧树脂成分100质量%中为20质量%以下。
[0038]
作为双酚a型环氧树脂的市售品,可举出“jer(注册商标)”825、“jer(注册商标)”826、“jer(注册商标)”827、“jer(注册商标)”828、“jer(注册商标)”834、“jer(注册商标)”1001、“jer(注册商标)”1002、“jer(注册商标)”1003、“jer(注册商标)”1004、“jer(注册商标)”1004af、“jer(注册商标)”1007、“jer(注册商标)”1009(以上三菱化学(株)制)、“epiclon(注册商标)”850(dic(株)制)、
“エポトート
(注册商标)”yd-128(新日铁住金化学(株)制)、“der(注册商标)
”‑
331、“der(注册商标)
”‑
332(
ダウケミカル
社制)等。
[0039]
作为双酚s型环氧树脂的市售品,可举出“epiclon(注册商标)”exa-1515(dic(株)制)等。
[0040]
本发明中的成分[c]为4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺)。成分[c]是为了向增强纤维基材的树脂注入中的树脂粘度长时间保持低粘度,向环氧树脂固化物和纤维增强复合材料给予高机械特性而需要的成分。作为这样的4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺)的市售品,可举出
“ロンザキュア
(注册商标)”m-cdea(
ロンザ
(株)制)等。
[0041]
本发明中的成分[c]优选在全部固化剂成分100质量中包含60质量%以上且90质量%以下。在全部固化剂成分100质量%中,包含成分[c]60质量%以上的情况下,易于适度抑制环氧树脂组合物的反应性,向增强纤维基材的树脂注入中的树脂粘度特别易于长时间保持低粘度。此外,在包含成分[c]90质量%以下的情况下,在加热固化时的升温速度慢的情况下将增强纤维基材连结的粘合剂在树脂中不过度熔融,易于保持粘合剂形状,因此在纤维增强复合材料中易于均匀地确保能够进行充分的塑性变形的层间厚度,易于表现充分的冲击后压缩强度。从这样的观点考虑,成分[c]的含量更优选在全部固化剂成分100质量%中为70质量%以上且90质量%以下的范围内。
[0042]
本发明中的成分[d]为4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺)。成分[d]是为了向环氧树脂固化物和纤维增强复合材料给予高耐热性和机械特性而需要的成分。作为这样的4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺)的市售品,可举出
“ロンザキュア
(注册商标)”m-dipa(
ロンザ
(株)制)等。
[0043]
本发明中的成分[d]优选在全部固化剂成分100质量%中包含5质量%以上且40质量%以下。在全部固化剂成分100质量%中包含成分[d]5质量%以上的情况下,易于表现高耐热性。此外,在包含40质量%以下的情况下,在加热固化时的升温速度慢的情况下将增强纤维基材连结的粘合剂表层在树脂中熔融,树脂与粘合剂的粘接性易于变得良好,因此易于表现充分的压缩强度、冲击后压缩强度、耐微裂纹性。从这样的观点考虑,成分[d]的含量更优选在全部固化剂成分100质量%中为5质量%以上且30质量%以下的范围内。
[0044]
本发明的纤维增强复合材料用环氧树脂组合物优选进一步包含[e]4,4
’‑
亚甲基双(2-异丙基-6-甲基苯胺)。以下,有时将[e]4,4
’‑
亚甲基双(2-异丙基-6-甲基苯胺)简称为成分[e]。通过包含成分[e],从而环氧树脂固化物和纤维增强复合材料易于获得更高的耐热性和更高的机械特性。作为这样的4,4
’‑
亚甲基双(2-异丙基-6-甲基苯胺)的市售品,可举出
“ロンザキュア
(注册商标)”m-mipa(
ロンザ
(株)制)等。
[0045]
本发明的纤维增强复合材料用环氧树脂组合物包含成分[e]的情况下的含量优选在全部固化剂成分100质量%中为5质量%以上且20质量%以下。在全部固化剂成分100质量%中,包含成分[e]5质量%以上的情况下,纤维增强复合材料的耐热性和压缩强度更易于提高。此外,在包含20质量%以下的情况下,由于即使将溶解了成分[e]的均匀的环氧树脂组合物长期冷冻保存,成分[e]也不易析出,因此操作性优异。此外,易于表现180℃下的高温时的高速固化性。从这样的观点考虑,成分[e]的含量更优选在全部固化剂成分100质量%中为5质量%以上且15质量%以下。
[0046]
此外,本发明的纤维增强复合材料用环氧树脂组合物可以包含具有能够与环氧树脂反应的活性基的化合物作为除成分[c]、成分[d]、成分[e]以外的固化剂。作为能够与环氧树脂反应的活性基,可举出例如,氨基、酸酐基等。环氧树脂组合物的保存稳定性越高越优选,一般而言液态的固化剂反应性高,因此除成分[c]、成分[d]、成分[e]以外的固化剂优选在室温下为固体。
[0047]
除成分[c]、成分[d]、成分[e]以外的固化剂优选为芳香族胺。此外,从耐热性、和机械特性的观点考虑,除成分[c]、成分[d]、成分[e]以外的固化剂更优选在分子内具有1~4个苯基。进一步,从通过赋予分子骨架的弯曲性从而树脂弹性模量提高,可以有助于机械特性提高考虑,进一步优选为环氧树脂的固化剂的骨架所包含的至少1个苯基为在邻位或间位具有氨基的苯基的芳香族多胺化合物。
[0048]
如果举出芳香族多胺化合物的具体例,则可举出间苯二胺、二氨基二苯基甲烷、二氨基二苯基砜、间苯二甲胺、二苯基-对二苯胺、它们的烷基取代体等各种衍生物、氨基的位置不同的异构体等。这些固化剂可以单独或并用2种以上。其中,从对环氧树脂固化物给予高耐热性方面考虑优选二氨基二苯基甲烷、二氨基二苯基砜。
[0049]
作为芳香族多胺化合物的固化剂的市售品,可举出
セイカキュア
s(和歌山精化工业(株)制)、mda-220(三井化学(株)制)、“jer
キュア
(注册商标)”w(三菱化学(株)制)、和3,3
’‑
das(三井化学(株)制)、
“ロンザキュア
(注册商标)”m-dea(
ロンザ
(株)制)、
“カヤハード
(注册商标)”a-a(pt)(日本化药(株)制)和
“ロンザキュア
(注册商标)”detda 80(
ロンザ
(株)制)等。
[0050]
此外,本发明的纤维增强复合材料用环氧树脂组合物可以根据需要包含固化促进剂、增塑剂、染料、颜料、无机填充材料、抗氧化剂、紫外线吸收剂、偶联剂、表面活性剂等作为其它成分。
[0051]
本发明的纤维增强复合材料用环氧树脂组合物只要是在即将注入含浸于增强纤维基材前包含成分[a]、成分[b]、成分[c]、成分[d]的组合物即可,可以在保存时作为包含全部成分的单体的组合物而保存,也可以作为包含任意选择的成分的多个组合物而保存。在作为多个组合物而保存的情况下,例如,也能够作为包含成分[a]和成分[b]的环氧主剂液、与包含成分[c]和成分[d]的固化剂组的2个组合物而保存,在即将注入含浸于增强纤维基材前,将环氧主剂液与固化剂组混合而调制包含全部成分的组合物而使用。在作为多个组合物而保存的情况下,各组合物中包含的成分的组合能够任意选择。在作为多个组合物而保存的情况下作为各组合物所包含的成分的组合,从防止由固化反应引起的增稠的观点考虑,优选将作为环氧树脂的成分[a]和成分[b]、与作为固化剂的成分[c]和成分[d]包含在分开的组合物中。
[0052]
在本发明中,作为环氧树脂所包含的环氧基总数(e)与固化剂中包含的胺化合物的活性氢总数(h)之比的h/e优选为1.1以上且1.4以下。h/e更优选为1.1以上且1.3以下。在h/e为1.1以上的情况下,易于获得环氧树脂固化物的塑性变形能力提高的效果。此外,在h/e为1.4以下的情况下,易于表现高耐热性。
[0053]
在本发明的纤维增强复合材料用环氧树脂组合物中,从本发明的效果的表现显著考虑,成分[a]~成分[d]的合计含量优选为70~100质量%,更优选为80~100质量%。
[0054]
本发明的纤维增强复合材料用环氧树脂组合物可以包含核壳橡胶粒子。核壳橡胶粒子在易于向纤维增强复合材料给予高韧性方面是优异的。这里所谓核壳橡胶粒子,是指将以交联橡胶等聚合物作为主成分的粒子状的核部分、与不同于核部分的聚合物通过接枝聚合等方法而被覆了核表面的一部分或整体的粒子。
[0055]
作为构成上述核壳橡胶粒子的核部分的成分,可举出由选自共轭二烯系单体、丙烯酸酯系单体、甲基丙烯酸酯系单体中的1种或多种聚合而成的聚合物、或有机硅树脂等。
2611、exl-3387(rohm&haas社制)等。此外,也可以使用
スタフィロイド
im-601、im-602(以上
ガンツ
化成(株)制)等将玻璃化转变温度为室温以上的玻璃状聚合物的核层用tg低的橡胶状聚合物的中间层覆盖,进一步将其周围用壳层覆盖了的具有3层结构的核壳橡胶粒子。通常,这些核壳橡胶粒子将以块状取出的物质粉碎而作为粉体进行操作,往往使粉体状核壳橡胶再次分散在热固性树脂组合物中。然而,对于该方法,具有难以使粒子在没有凝集的状态,即一次粒子的状态下稳定地分散这样的问题。针对该问题,通过使用从核壳橡胶粒子的制造过程一次也不以块状取出,最终可以以在热固性树脂的一成分、例如环氧树脂中以一次粒子分散了的母料的状态进行操作的物质,从而可以获得优选的分散状态。可以以这样的母料的状态进行操作的核壳橡胶粒子例如可以通过日本特开2004-315572号公报的实施例1~3中任一实施例所记载的方法来制造。对于该制造方法,首先,将核壳橡胶使用以乳液聚合、分散聚合、悬浮聚合为代表的在水介质中进行聚合的方法而获得核壳橡胶粒子分散了的悬浮液。接下来,在这样的悬浮液中混合与水显示部分溶解性的有机溶剂例如丙酮、甲基乙基酮等酮系溶剂、四氢呋喃、二烷等醚系溶剂后,使水溶性电解质例如氯化钠、氯化钾接触,使有机溶剂层与水层进行相分离,将水层分离除去,获得所得的核壳橡胶粒子分散了的有机溶剂。然后,在混合了环氧树脂后,将有机溶剂蒸发除去,获得核壳橡胶粒子在环氧树脂中以一次粒子的状态分散了的母料。作为通过这样的方法而制造出的核壳橡胶粒子分散环氧母料,可以使用由(株)
カネカ
市售的
“カネエース
(注册商标)”。
[0061]
包含成分[f]的情况下的成分[f]的含量优选相对于全部环氧树脂成分100质量份为1质量份以上且10质量份以下,更优选为1质量份以上且8质量份以下。在为1质量份以上的情况下,易于获得高韧性的环氧树脂固化物。此外,在为10质量份以下的情况下,易于获得高弹性模量的环氧树脂固化物,进一步树脂中的核壳橡胶粒子的分散性也易于变得良好。需要说明的是,即使在核壳橡胶粒子具有环氧基的情况下,核壳橡胶粒子也不相当于环氧树脂成分。
[0062]
作为在纤维增强复合材料用环氧树脂组合物中混合核壳橡胶粒子的方法,可以使用一般使用的分散方法。可举出使用例如三辊、球磨机、珠磨机、喷射磨机、均化器、自转/公转混合机等的方法。此外,也可以优选使用混合上述核壳橡胶粒子分散环氧母料的方法。然而,有时即使以一次粒子的状态分散,也因过度的加热、粘度的降低而发生再凝集。因此,在核壳橡胶粒子的分散/混配、和分散后与其它成分混合/混炼的情况下,优选在核壳橡胶粒子的再凝集不发生的温度/粘度的范围进行。具体而言,虽然根据组合物而不同,但是例如,在150℃以上的温度下混炼了的情况下,具有组合物的粘度下降而发生凝集的可能性,因此优选在低于其的温度下混炼。然而,关于在固化工艺中达到150℃以上的情况,在升温时随着凝胶化而妨碍再凝集,因此可以超过150℃。
[0063]
本发明的纤维增强复合材料用环氧树脂组合物优选通过采用e型粘度计的等温测定而获得的、120℃恒定240分钟后的树脂粘度η(mpa
·
s)满足20≤η≤200。在η为20mpa
·
s以上的情况下,树脂注入温度下的粘度不过低,在向增强纤维基材的注入时由卷入空气而产生的坑引起的未含浸不易发生。此外,如果在树脂注入温度下,环氧树脂组合物的反应性高,则有时在注入过程中粘度增加,含浸性降低,产生未含浸部,或成型花费时间等,成型变得困难,但在η为200mpa
·
s以下的情况下,由于树脂注入温度下的粘度充分低,因此向增强纤维基材的含浸性良好,未含浸不易发生。从这样的观点考虑,更优选满足20≤η≤180。需
要说明的是,本发明中的粘度按照jis z8803(1991)中的“円
すい
-板形回転粘度計
による
粘度測定方法(采用圆锥-板形旋转粘度计的粘度测定方法)”,使用装备了标准锥形转子(1
°
34
’×
r24)的e型粘度计(东京计器(株)制,tve-30h),以旋转速度50转/分钟测定。
[0064]
本发明的纤维增强复合材料用环氧树脂组合物优选从70℃以升温速度0.5℃/分钟进行了加热时的凝胶化温度在170℃以上且185℃以下的范围内。这里所谓环氧树脂组合物的凝胶化,是指树脂中的环氧树脂与固化剂的反应进行,流动性消失。具体而言,使用atd-1000(alpha technologies(株)制)等热固化测定装置从70℃以升温速度0.5℃/分钟加热而进行环氧树脂组合物的动态粘弹性测定,将由伴随固化反应进行的转矩上升而求出的复数粘性率达到1.0
×
105pa
·
s时的温度设为凝胶化温度。在凝胶化温度为170℃以上的情况下,凝胶化温度易于高于将增强纤维基材连结的粘合剂的熔点。在凝胶化温度高于上述粘合剂的熔点的情况下,在低于环氧树脂组合物的凝胶化温度的温度下将增强纤维基材连结的粘合剂熔化出,因此环氧树脂组合物进入到变松了的聚酰胺分子链的间隙,在该状态下环氧树脂经过凝胶化而固化,从而树脂与聚酰胺分子链缠绕而界面粘接强度提高,压缩强度、耐冲击性、耐微裂纹性易于提高。此外,在凝胶化温度为185℃以下的情况下,凝胶化温度相对于将增强纤维基材连结的粘合剂的熔点不易过高。在那样的情况下,将增强纤维基材连结的粘合剂在树脂中不会过度熔融,保持粘合剂形状,因此在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度,易于表现充分的冲击后压缩强度。从这样的观点考虑,凝胶化温度更优选为175℃以上且185℃以下的范围内。
[0065]
本发明的纤维增强复合材料用环氧树脂组合物优选在180℃下进行120分钟固化而得的环氧树脂固化物的玻璃化转变温度tg为170℃以上且190℃以下。纤维增强复合材料的耐热性依赖于将环氧树脂组合物固化而成的环氧树脂固化物的玻璃化转变温度。通过使tg为170℃以上,从而环氧树脂固化物的耐热性易于被确保。此外,通过为190℃以下,从而环氧树脂组合物的固化收缩被抑制,而且,易于防止由环氧树脂组合物与增强纤维的热膨胀的不同产生的纤维增强复合材料的表面品质的恶化。此外,根据耐热性与表面品质的关系,更优选玻璃化转变温度tg为175℃以上且190℃以下。这里,将环氧树脂组合物固化而成的环氧树脂固化物的玻璃化转变温度tg通过使用了动态粘弹性测定(dma)装置的测定而求出。即,使用从树脂固化板切出了的矩形的试验片,进行升温下dma测定,将所得的储能弹性模量g’的拐点的温度设为tg。测定条件如实施例所记载的那样。
[0066]
本发明的纤维增强复合材料包含本发明的纤维增强复合材料用环氧树脂组合物的固化物、和增强纤维基材。
[0067]
本发明的纤维增强复合材料例如可以通过将由环氧树脂和固化剂构成的环氧树脂组合物注入到配置在加热了的成型模内的增强纤维基材中,使其含浸,在该成型模内进行固化来获得。作为其具体的成型方法,如上述那样,从生产性、所得的成型体的形状自由度这样的观点考虑,适合使用rtm法。此外,在制造这样的纤维增强复合材料的方法中,成型模使用具有多个注入口的物质,将环氧树脂组合物从多个注入口同时,或设置时间差而依次注入等,根据想要获得的纤维增强复合材料来选择适当的条件由于可获得可以与各种形状、大小的成型体对应的自由度因此是优选的。对这样的注入口的数目、形状没有限制,但为了能够以短时间进行注入,注入口越多越好,其配置优选为根据成型品的形状而可以使树脂的流动长度短的位置。
[0068]
作为纤维增强复合材料所使用的增强纤维基材,往往使用:使用热熔性的粘合剂(增粘剂)将增强纤维机织物等片状基材叠层、赋形,加工为与所希望的制品接近的形状的预成型件。作为热熔性的粘合剂,热塑性树脂和热固性树脂都能够应用。作为粘合剂的形态,没有特别限定,可以采用膜、带、长纤维、短纤维、细纱、机织物、针织物、无纺织物、网状体、粒子等形态。其中,粒子形态、或无纺织物形态可以特别适合使用。需要说明的是,将粘合剂为粒子形态的情况称为粘合剂粒子,将粘合剂为无纺织物形态的情况称为粘合剂无纺织物。
[0069]
在采用粒子形态作为粘合剂的形态的情况下,其平均粒径优选为10μm以上且500μm以下。这里平均粒径是指中间粒径。粘合剂粒子的平均粒径例如可以使用激光衍射型粒度分布计等而测定。如果平均粒径为10μm以上,则制成预成型件时的粘接强度和作业性易于提高。从这样的观点考虑,平均粒径更优选为30μm以上。如果平均粒径为500μm以下,则在制成预成型件时增强纤维不易发生起伏,所得的纤维增强复合材料的力学特性易于提高。从这样的观点考虑,平均粒径更优选为300μm以下。
[0070]
在采用无纺织物形态作为粘合剂的形态的情况下,构成无纺织物的纤维的平均直径优选为10μm以上且300μm以下。这里平均直径是利用扫描型电子显微镜观察粘合剂无纺织物的截面,关于任意选择的100个纤维而测定直径的长度,算出了其算术平均值的值。在纤维的截面形状不是正圆的情况下,将短径作为其直径而测定。如果平均直径为10μm以上,则预成型件的粘接强度易于提高。如果平均直径为300μm以下,则预成型件的增强纤维不易发生起伏,所得的纤维增强复合材料的力学特性易于提高。从这样的观点考虑,平均直径更优选为100μm以下。
[0071]
使粘合剂附着在增强纤维基材的至少表面而作为带有粘合剂的增强纤维基材而使用。此外,带有粘合剂的增强纤维基材在至少表面具有上述粘合剂,被使用于预成型件。
[0072]
在本发明的纤维增强复合材料中,优选上述增强纤维基材通过无纺织物形态的粘合剂而被连结而成的预成型件。通过为增强纤维基材通过无纺织物形态的粘合剂而被连结而成的预成型件,从而能够在基材上均匀地配置粘合剂,因此可确保环氧树脂组合物的含浸流路。因此,含浸性特别优异,空隙极其不易发生。此外,即使与粒子形态的粘合剂相比粘合剂的附着量少,也可以同等地维持制成预成型件时的形态固定的效果,在制成纤维增强复合材料时易于表现基体树脂固有的高耐热性、力学特性。
[0073]
本发明的纤维增强复合材料优选纤维增强复合材料用环氧树脂组合物的固化物中的纤维增强复合材料用环氧树脂组合物从70℃以升温速度0.5℃/分钟进行了加热时的凝胶化温度为将增强纤维基材连结的粘合剂的熔点以上。凝胶化温度与熔点之差(凝胶化温度-熔点)更优选为1℃以上,进一步优选为4℃以上。此外,凝胶化温度与熔点之差优选为20℃以下,进一步优选为15℃以下。在上述温度差的范围内的情况下,在低于环氧树脂组合物的凝胶化温度的温度下将增强纤维基材连结的粘合剂熔化出,因此环氧树脂组合物进入到变松了的聚酰胺分子链的间隙,在该状态下环氧树脂经过凝胶化而固化,从而树脂与聚酰胺分子链缠绕而界面粘接强度提高,压缩强度、耐冲击性、耐微裂纹性易于提高,并且,将增强纤维基材连结的粘合剂在树脂中不会过度熔融,保持粘合剂形状,因此在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度,易于表现充分的冲击后压缩强度。
[0074]
在本发明的纤维增强复合材料中,优选无纺织物形态的粘合剂由熔点为165℃以上且180℃以下的聚酰胺构成。在聚酰胺的熔点为165℃以上的情况下,在固化时可以保持形态,易于均匀地确保能够进行充分的塑性变形的层间厚度,或由于无纺织物为连续相,因此可以易于有效率地阻断裂纹,易于表现耐冲击性。在聚酰胺的熔点为180℃以下的情况下,聚酰胺的熔点易于低于环氧树脂组合物的凝胶化温度。在聚酰胺的熔点低于环氧树脂组合物的凝胶化温度的情况下,由于在低于环氧树脂组合物的凝胶化温度的温度下聚酰胺熔化出,因此环氧树脂组合物进入到变松了的聚酰胺分子链的间隙,在该状态下环氧树脂经过凝胶化而固化,从而有时树脂与聚酰胺分子链缠绕而界面粘接强度提高,压缩强度、耐冲击性、耐微裂纹性更提高。
[0075]
在本发明的纤维增强复合材料中,优选在纤维增强基材的一面或两面附着有上述无纺织物形态的粘合剂。
[0076]
在本发明的纤维增强复合材料中,优选在纤维增强基材的表面附着的上述无纺织物形态的粘合剂的附着量为每一面0.5g/m2以上且10g/m2以下。上述附着量更优选为1g/m2以上且7g/m2以下。在附着量为0.5g/m2以上的情况下,制成预成型件时的形态固定易于变得容易。此外,在为10g/m2以下的情况下,基体树脂易于显示充分的含浸性,不易产生空隙。此外,如上述那样,在采用无纺织物形态作为粘合剂的形态的情况下,即使与采用粒子形态的情况相比粘合剂的附着量少,也可以维持同等的效果。具体而言,使粒子形态等的通常的粘合剂附着于表面的情况下的附着量优选为每一面0.5g/m2以上且50g/m2以下,优选为1g/m2以上且30g/m2以下的附着量,与此相对,对于无纺织物形态的粘合剂,也能够在同等地维持制成预成型件时的形态固定的效果的同时,使附着量为0.5g/m2以上且10g/m2以下。
[0077]
作为获得预成型件的方法,可举出例如,将至少表面具有上述粘合剂的带有粘合剂的增强纤维基材叠层,将形态固定的方法。更具体而言,例如,使粘合剂通过加热而附着在增强纤维基材的至少一面的至少表面而制成带有粘合剂的增强纤维基材后,将其多片叠层,从而获得至少在叠层的层间具有粘合剂的叠层体。可举出通过将其进行加热和冷却,粘合剂在基材层间固着而将形态固定,获得至少在叠层的层间具有粘合剂的预成型件的方法。
[0078]
通常,预成型件可以将粘合剂附着了的带有粘合剂的增强纤维基材切出为规定的形状,在模上叠层,施加适当的热和压力而制作。加压的手段既可以使用压机,也可以使用将用真空袋膜包围而将内部用真空泵抽吸通过大气压进行加压的方法。
[0079]
本发明的纤维增强复合材料的增强纤维的纤维体积含有率优选为45%以上且70%以下的范围内,更优选为50%以上且65%以下的范围内。在纤维体积含有率为45%以上的情况下,可获得进一步高弹性模量且轻量化效果优异的纤维增强复合材料。此外,在70%以下的情况下,没有由增强纤维彼此的擦蹭引起的强度降低,可获得进一步抗拉强度等力学特性优异的纤维增强复合材料。
[0080]
构成本发明中的增强纤维基材的增强纤维没有特别限定,可举出玻璃纤维、碳纤维、石墨纤维、芳族聚酰胺纤维、硼纤维、氧化铝纤维和碳化硅纤维等。可以将这些增强纤维混合使用2种以上。其中,为了获得更轻量,耐久性更高的纤维增强复合材料,优选使用碳纤维、石墨纤维。特别是,在材料的轻量化、高强度化的要求高的用途中,从具有优异的比弹性模量和比强度考虑,优选在本发明的纤维增强复合材料中,构成增强纤维基材的增强纤维
为碳纤维。
[0081]
作为碳纤维,能够根据用途而使用所有种类的碳纤维,但从耐冲击性方面考虑,优选为拉伸弹性模量具有230gpa以上且400gpa以下的拉伸弹性模量的碳纤维。此外,从强度的观点考虑,从获得具有高刚性和机械强度的复合材料考虑,优选为抗拉强度为4.4gpa以上且6.5gpa以下的碳纤维。此外,拉伸伸长率也是重要的要素,优选为1.7%以上且2.3%以下的高强度高伸长率碳纤维。因此,兼备拉伸弹性模量至少为230gpa,抗拉强度至少为4.4gpa,拉伸伸长率至少为1.7%这样的特性的碳纤维是最适合的。
[0082]
作为碳纤维的市售品,可举出
“トレカ
(注册商标)”t800g-24k、
“トレカ
(注册商标)”t800s-24k、
“トレカ
(注册商标)”t700g-24k、
“トレカ
(注册商标)”t300-3k、和
“トレカ
(注册商标)”t700s-12k(以上東
レ
(株)制)等。
[0083]
本发明的纤维增强复合材料如上述那样,包含本发明的纤维增强复合材料用环氧树脂组合物的环氧树脂固化物、和增强纤维基材。在纤维增强复合材料特别是在航空器领域中使用的情况下,要求高耐热性、力学特性。本发明的纤维增强复合材料优选作为基体树脂的环氧树脂固化物的玻璃化转变温度为170℃以上且190℃以下。通过具有这样的范围的玻璃化转变温度,从而耐热性优异,并且反映环氧树脂固化物所具有的高机械特性。因此,本发明的纤维增强复合材料的作为湿热时的0
°
压缩强度的h/w0
°
压缩强度高,优选为1000mpa以上,在更优选的方案中可以显示1100mpa以上这样的高h/w0
°
压缩强度。此外,冲击后压缩强度可以显示优选260mpa以上、更优选270mpa以上这样的高冲击后压缩强度。
[0084]
本发明中的纤维增强复合材料的耐微裂纹性优异。这里所谓微裂纹,是有时在航空器用途中被使用的纤维增强复合材料中产生的数十μm左右的微小的裂纹,已知如果在反复进行从70℃左右的高温到-50℃左右的低温的温度变化的环境中多次暴露则易于产生。如果纤维增强复合材料内的基体树脂被暴露在70℃左右的高温到-50℃左右的低温的气氛下则基体树脂本身想要收缩,但由于周围通过几乎不收缩的增强纤维包围因此不能收缩,作为结果,在基体树脂本身的内部拉伸的应力内在(热残余应力),如果其被反复进行则有时环境疲劳蓄积,在纤维增强复合材料的内部产生微裂纹。该现象特别是在富树脂部中显著,微裂纹也往往在富树脂部产生。如果在产生了微裂纹的状态下进一步施加环境疲劳,则可能微裂纹向更巨大的裂纹进一步生长,最后通过该巨大的裂纹而使纤维增强复合材料的力学特性降低。由于rtm法的限制,在成型时需要树脂的流路,因此难以将完全没有富树脂部的纤维增强复合材料成型,为了防止因环境疲劳而产生的微裂纹而提高基体树脂固化物的破坏韧性、和以避免在基体树脂产生的残余热应力过高的方式适度地抑制弹性模量是有效果的。
[0085]
在本发明的纤维增强复合材料中,从纤维增强复合材料的长期耐久性这样的观点考虑,微裂纹数优选小于10个,更优选为5个以下。微裂纹数的具体的算出方法如后述那样。
[0086]
本发明的纤维增强复合材料的制造方法是将加热到50℃以上且130℃以下的本发明的纤维增强复合材料用环氧树脂组合物注入到配置在加热到90℃以上且180℃以下的成型模内的增强纤维基材中,使其含浸,在该成型模内进行固化。从向增强纤维基材的含浸性方面考虑,纤维增强复合材料用环氧树脂组合物基于环氧树脂组合物的初始粘度与粘度上升的关系,在注入前加热到选自50℃以上且130℃以下的范围中的温度。此时,如果在注入温度下,环氧树脂组合物的反应性高,则有时在注入过程中粘度增加而成型变得困难。因
此,本发明的纤维增强复合材料用环氧树脂组合物在120℃恒定240分钟后的树脂粘度优选为200mpa
·
s以下,更优选为180mpa
·
s以下。需要说明的是,在本发明中粘度测定通过后述方法进行。如果从注入开始起240分钟后的粘度高于上述范围则有时环氧树脂组合物的含浸性变得不充分。此外,120℃恒定240分钟后的树脂粘度优选为20mpa
·
s以上,树脂注入温度下的粘度不会过低,可以防止在向增强纤维基材的注入时由卷入空气而产生的坑引起的未含浸。此外,成型模温度优选为90℃以上且180℃以下。通过使成型模温度为90℃以上且180℃以下,从而将固化所需要的时间缩短,同时使脱模后的热收缩缓和,从而可以获得表面品质良好的纤维增强复合材料。
[0087]
纤维增强复合材料用环氧树脂组合物的注入压力通常为0.1mpa以上且1.0mpa以下。也可以使用将模内抽真空而注入环氧树脂组合物的vartm(真空辅助树脂传递模塑,vacuum assist resins transfer molding)法。从注入时间与设备的经济性方面考虑,纤维增强复合材料用环氧树脂组合物的注入压力优选为0.1mpa以上且0.6mpa。此外,在进行加压注入的情况下,如果在注入纤维增强复合材料用环氧树脂组合物前将模内抽成真空,则也抑制空隙的产生,是优选的。
[0088]
本发明的纤维增强复合材料由于具有优异的耐热性、高压缩强度、耐冲击性、耐久性,因此可以优选用于机身、主翼、尾翼、动翼、整流板、整流罩、门、座位、内装材料等航空器构件、电动机壳体、主翼等宇宙飞船构件、结构体、天线等人造卫星构件、外板、底盘、空气动力构件、座位等汽车构件、结构体、座位等铁道车辆构件、船体、座位等船舶构件等大量结构材料。
[0089]
实施例
[0090]
以下,通过实施例对本发明的纤维增强复合材料用环氧树脂组合物等进一步详细地说明,但本发明不限定于它们。
[0091]
<树脂原料>
[0092]
为了获得各实施例/比较例的环氧树脂组合物,使用了以下树脂原料。需要说明的是,表中的环氧树脂组合物的栏中的各成分的数值表示含量,其单位只要没有特别指明就是“质量份”。
[0093]
1.[a]四缩水甘油基二氨基二苯基甲烷
[0094]
·
“アラルダイト
(注册商标)”my721(n,n,n’,n
’‑
四缩水甘油基-4,4
’‑
二氨基二苯基甲烷,环氧当量:113g/mol,
ハンツマン
·
ジャパン
(株)制)
[0095]
2.[b]双酚f型环氧树脂
[0096]
·“epiclon(注册商标)”830(双酚f的二缩水甘油基醚,环氧当量:170g/mol,dic(株)制)
[0097]
·
“アラルダイト
(注册商标)”gy282(双酚f的二缩水甘油基醚,环氧当量:171g/mol,
ハンツマン
·
ジャパン
(株)制)
[0098]
3.除成分[a]、成分[b]以外的环氧树脂
[0099]
·“epiclon(注册商标)”850(双酚a的二缩水甘油基醚,环氧当量:188g/mol,dic(株)制)。
[0100]
4.[c]4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺)
[0101]
·
“ロンザキュア
(注册商标)”m-cdea(4,4
’‑
亚甲基双(3-氯-2,6-二乙基苯胺,活
性氢当量:95g/mol,
ロンザ
(株)制)。
[0102]
5.[d]4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺)
[0103]
·
“ロンザキュア
(注册商标)”m-dipa(4,4
’‑
亚甲基双(3,3’,5,5
’‑
四异丙基苯胺),活性氢当量:93g/mol,
ロンザ
(株)制)
[0104]
6.[e]4,4
’‑
亚甲基双(2-异丙基-6-甲基苯胺)
[0105]
·
“ロンザキュア
(注册商标)”m-mipa(3,3
’‑
二异丙基-5,5
’‑
二甲基-4,4
’‑
二氨基二苯基甲烷,活性氢当量:78g/mol,
ロンザ
(株)制)
[0106]
7.除成分[c]、成分[d]、成分[e]以外的固化剂
[0107]
·
“ロンザキュア
(注册商标)”m-dea(3,3’,5,5
’‑
四乙基-4,4
’‑
二氨基二苯基甲烷,活性氢当量:78g/mol,
ロンザ
(株)制)
[0108]
·
“カヤハード
(注册商标)”a-a(pt)(以2,2
’‑
二乙基-4,4
’‑
二氨基二苯基甲烷作为主成分的混合物,活性氢当量:64g/mol,液态,日本化药(株)制)
[0109]
8.添加剂
[0110]
·
“カネエース
(注册商标)”mx-416(
“アラルダイト
(注册商标)”my721:75质量%/核壳橡胶粒子(体积平均粒径:100nm,核部分:交联聚丁二烯,壳部分:甲基丙烯酸甲酯/甲基丙烯酸缩水甘油酯/苯乙烯共聚聚合物):25质量%的母料,(株)
カネカ
制)
[0111]
·
“スタフィロイド
(注册商标)”ac-3355(核壳橡胶粒子(体积平均粒径:500nm,核部分:交联聚丙烯酸丁酯,壳部分:交联聚苯乙烯,
アイカ
工業(株)制)。
[0112]
<环氧树脂组合物的调制>
[0113]
以表所记载的含有比例将各成分混合,调制出环氧树脂组合物。
[0114]
<树脂固化板的制作>
[0115]
在将上述调制出的环氧树脂组合物在减压下进行了脱泡后,注入到通过2mm厚的
“テフロン
(注册商标)”制间隔物以成为厚度2mm的方式设定的模型中。在180℃的温度下使其固化120分钟,获得了厚度2mm的树脂固化板。
[0116]
<粘合剂的制作>
[0117]
按照下述制造方法而制作出粘合剂。
[0118]
(粘合剂1的制作)
[0119]
将己内酰胺1.2g、十二内酰胺18.8g、离子交换水10g加入压力容器中进行密闭,进行了氮气置换。开始加热,在罐内压力达到10kg/cm2后,一边使水分放出到体系外一边将罐内压力以10.0kg/cm2保持,如果内温变为170℃,则经50分钟将罐内压力恢复到常压,进一步在氮气流下使其反应2小时而完成了聚合。然后,从压力容器将聚合物条状地排出而制成粒状,将其在80℃下真空干燥24小时,获得了尼龙6/12(=6/94质量%)的共聚物pa-1。将从设置了1个孔口的口模排出了的pa-1(熔点:170℃)的纤维,使用前端设置了冲击板的抽吸器和压缩空气而进行了拉伸后,散布在金属网上而进行了捕集。将在金属网上捕集而成的纤维片使用加热加压机进行热粘接,制作出无纺织物形态的粘合剂1。
[0120]
(粘合剂2的制作)
[0121]
将从设置了1个孔口的模排出了的pes(住友化学(株)制
“スミカエクセル
(注册商标)”5003p,熔点:无)的纤维,使用前端设置了冲击板的抽吸器和压缩空气而进行了拉伸后,散布在金属网上而进行了捕集。将在金属网上捕集而成的纤维片使用加热加压机进行
热粘接,制作出无纺织物形态的粘合剂2。
[0122]
(粘合剂3的制作)
[0123]
将从设置了1个孔口的口模排出了的pa-2(東
レ
(株)制
“アミラン
(注册商标)”cm1001,熔点:225℃)的纤维,使用前端设置了冲击板的抽吸器和压缩空气而进行了拉伸后,散布在金属网上而进行了捕集。将在金属网上捕集而成的纤维片使用加热加压机进行热粘接,制作出无纺织物形态的粘合剂3。
[0124]
(粘合剂4的制作)
[0125]
将从设置了1个孔口的口模排出了的pa-3(
アルケマ
(株)制“platamid(注册商标)”m1657,熔点:110℃)的纤维,使用前端设置了冲击板的抽吸器和压缩空气而进行了拉伸后,散布在金属网上而进行了捕集。将在金属网上捕集而成的纤维片使用加热加压机进行热粘接,制作出无纺织物形态的粘合剂4。
[0126]
(粘合剂5的制作)
[0127]
将甲酚酚醛清漆型环氧树脂(dic(株)制“epiclon(注册商标)”n-660)15质量份、双酚型环氧树脂(三菱
ケミカル
(株)制“jer(注册商标)”825)25质量份、聚醚砜(住友化学(株)制
“スミカエクセル
(注册商标)”pes5200p)60质量份在180℃的温度条件下使用小型双螺杆挤出机(s1krc捏合机,(株)栗本铁工所)进行混炼而调制出粘合剂树脂组合物。将调制出的粘合剂树脂组合物利用锤磨机(pulverizer,
ホソカワミクロン
(株)制),使用孔尺寸1mm的筛网,使用液氮进行冷冻粉碎而获得了粒子形态的粘合剂5。将这样的粒子过筛孔尺寸150μm和75μm的筛子,将残留在筛孔尺寸75μm的筛子上的粘合剂粒子用于评价。
[0128]
<带有粘合剂的增强纤维基材的制作>
[0129]
使所得的粘合剂附着在碳纤维单向机织物(平纹织物,经纱:碳纤维t800s-24k-10c東
レ
(株)制,碳纤维目付295g/m2,经纱密度7.2根/25mm,纬纱:玻璃纤维ece225 1/0 1z日东纺(株)制,纬纱密度7.5根/25mm)的一面。附着量在粘合剂1~粘合剂4的情况下设为5g/m2,在粘合剂5的情况下设为10g/m2。然后,使用远红外线加热器进行加热,使粘合剂熔合,获得了向单侧表面赋予了粘合剂的带有粘合剂的增强纤维基材。
[0130]
<评价>
[0131]
各实施例中的评价如以下那样进行。需要说明的是,测定n数只要没有特别指明,就是n=1。
[0132]
(1)环氧树脂组合物的凝胶化温度的测定
[0133]
将要测定的检体使用热固化测定装置atd-1000(alpha technologies(株)制),在加热到70℃的平台投入样品,一边以升温速度0.5℃/分钟升温,一边以频率1.0hz、应变1%进行动态粘弹性测定,求出了复数粘性率。此时,将复数粘性率达到1.0
×
105pa
·
s的温度设为凝胶化温度。需要说明的是,作为检体,使用了将各成分混合,搅拌1分钟后的环氧树脂组合物。
[0134]
(2)120℃240分钟保持后的环氧树脂组合物的粘度的测定
[0135]
将要测定的检体按照jis z8803(1991)中的“円
すい
-平板形回転粘度計
による
粘度測定方法(采用圆锥-平板形旋转粘度计的粘度测定方法)”,使用安装了标准锥形转子(1
°
34
’×
r24)的e型粘度计,在保持于120℃的状态下测定。作为e型粘度计,使用了东京计器(株)制tve-30h。需要说明的是,作为检体,使用了将各成分混合,搅拌1分钟后的环氧树
脂组合物。
[0136]
(3)环氧树脂固化物的玻璃化转变温度tg测定
[0137]
从树脂固化板切出宽度12.7mm、长度40mm的试验片,使用dma装置ares(ta
インスツルメンツ
社制)而进行了tg测定。测定条件为升温速度5℃/分钟。将在测定中获得的储能弹性模量g’的拐点的温度设为tg。
[0138]
(4)纤维增强复合材料的湿热时0
°
压缩强度测定
[0139]
在具有400mm
×
400mm
×
1.2mm的板状模腔的模具中,放置将切出为395mm
×
395mm的上述带有粘合剂的增强纤维基材以纤维方向为0
°
而沿0
°
方向拉齐而叠层了4片的物质,进行了合模。接着,在将模具加热到120℃后,将另行预先加热到120℃的环氧树脂组合物使用树脂注入装置,以注入压力0.2mpa注入到成型模内。在注入后,将模具从120℃以0.5℃/分钟升温直到180℃,在180℃下使其固化了2小时后,降温直到30℃而获得了纤维增强复合材料。
[0140]
从通过上述方法而获得的纤维增强复合材料,以0
°
方向与长度方向变得相同的方式切割宽度12.7mm、长度79.4mm的试验片,制作出0
°
压缩强度用试验片。关于该试验片,在72℃温水中浸渍了14天后,测定了纤维增强复合材料的0
°
压缩强度。0
°
压缩强度的测定按照astm d695,作为试验机,使用材料万能试验机(
インストロン
·
ジャパン
(株)制4208型
インストロン
),将测定时的十字头速度设为1.27mm/分钟,将测定温度设为82℃。
[0141]
(5)纤维增强复合材料的冲击后压缩强度测定
[0142]
在具有400mm
×
400mm
×
4.8mm的板状模腔的模具中,放置将切出为395mm
×
395mm的上述带有粘合剂的增强纤维基材以纤维方向为0
°
重复3次(45
°
/0
°
/-45
°
/90
°
)而叠层了12片后,重复3次(90
°
/-45
°
/0
°
/45
°
)而叠层了12片的物质,进行了合模。接着,在将模具加热到120℃后,将预先另行加热到120℃的环氧树脂组合物使用树脂注入装置,以注入压力0.2mpa注入到成型模内。在注入后,将模具从120℃以0.5℃/分钟升温直到180℃,在180℃下固化了2小时后,降温直到30℃而获得了纤维增强复合材料。
[0143]
从通过上述方法获得的纤维增强复合材料,以0
°
方向与长度方向变得相同的方式切割宽度101.6mm、长度152.4mm的试验片,按照sacma srm 2r-94,测定了冲击后压缩强度。作为试验机,使用了材料万能试验机(
インストロン
·
ジャパン
(株)制,1128型
テンシロン
)。这里,落锤冲击的能量设为6.7j/mm,十字头速度设为1.27mm/分钟。
[0144]
(6)纤维增强复合材料的微裂纹数
[0145]
从通过上述(5)而获得的纤维增强复合材料,以0
°
方向与长度方向变得相同的方式切割宽度75mm、长度50mm的试验片。将所得的试验片使用市售的恒温恒湿槽和环境试验机暴露于以下a、b、c的步骤所示的环境条件。
[0146]
a.使用市售的恒温恒湿槽,在49℃、95%/rh的环境暴露12小时。
[0147]
b.在暴露后,转移到市售的环境试验机,首先在-54℃的环境下暴露1小时。然后以10℃
±
2℃/分钟的升温速度使其升温直到71℃。在升温后在71℃下保持了5分钟
±
1分钟后,以10℃
±
2℃/分钟使其降温直到-54℃,在-54℃下保持5分钟
±
1分钟。将该从-54℃升温直到71℃并降温直到-54℃的循环定义为1循环,将该循环重复200次。
[0148]
c.将上述恒温恒湿槽中的环境暴露和环境试验机中的循环合并定义为1区块,重复5区块。
[0149]
从进行了上述环境暴露的试验片的纵向的距中央
±
10mm的区域切出宽度25mm,将切出面作为观察面进行研磨,使用市售的显微镜以200倍的倍率观察观察面,计测了产生的裂纹数。从纤维增强复合材料的长期耐久性这样的观点考虑,通过上述方法而观察到的微裂纹数优选小于10个,更优选为5个以下,因此将5个以下的情况设为a,将6个以上且小于10个的情况设为b,将10个以上的情况设为c。
[0150]
(实施例1)
[0151]
如表1-1所示那样,加入作为成分[a]的
“アラルダイト
(注册商标)”my721 70质量份、作为成分[b]的“epiclon(注册商标)”830 30质量份、作为成分[c]的
“ロンザキュア
(注册商标)”m-cdea 59质量份、作为成分[d]的
“ロンザキュア
(注册商标)”m-dipa 25质量份,在80℃下搅拌1小时而调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为177℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为140mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为175℃,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1060mpa,是良好的,冲击后压缩强度为280mpa,微裂纹数为5个以下,是特别良好的。
[0152]
(实施例2~5)
[0153]
将各成分的含量如表1-1所示那样变更了,除此以外,与实施例1同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为170℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的温度。其中,实施例2、3、5为175℃以上且185℃以下,是特别优选的结果。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为200mpa
·
s以下,向增强纤维的含浸性也良好。其中,实施例2、3、5为180mpa
·
s以下,是特别优选的结果。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为175℃以上,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,微裂纹数小于10个,力学特性、耐久性也良好。其中,实施例2、3、5中湿热时0
°
压缩强度为1100mpa以上,冲击后压缩强度为270mpa以上,微裂纹数为5个以下,是特别优选的结果。
[0154]
(实施例6)
[0155]
如表1-1所示那样,加入作为成分[a]的
“アラルダイト
(注册商标)”my721 80质量份、作为成分[b]的“epiclon(注册商标)”830 20质量份、作为成分[c]的
“ロンザキュア
(注册商标)”m-cdea 52质量份、作为成分[d]的
“ロンザキュア
(注册商标)”m-dipa 17质量份、作为成分[e]的
“ロンザキュア
(注册商标)”m-mipa 15质量份,在80℃下搅拌1小时而调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为170℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为200mpa
·
s,向增强纤维的含浸性也良好。此外,通过上述
方法而制作树脂固化物,测定了固化物的tg,结果为186℃,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1030mpa,冲击后压缩强度为260mpa,微裂纹数为6个以上且小于10个,是良好的。
[0156]
(实施例7、8)
[0157]
将各成分的含量如表1-1、表1-2所示那样变更了,除此以外,与实施例6同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为175℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为180mpa
·
s以下,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为175℃以上,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1100mpa以上,冲击后压缩强度为270mpa以上,微裂纹数为5个以下,特别是力学特性、耐久性也良好。
[0158]
(实施例9)
[0159]
作为成分[b],使用了
“アラルダイト
(注册商标)”gy282,除此以外,与实施例7同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为175℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为170mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为184℃,耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1150mpa,是良好的,冲击后压缩强度为270mpa,微裂纹数为5个以下,是特别良好的。
[0160]
(实施例10)
[0161]
代替成分[e],而使用了
“ロンザキュア
(注册商标)”m-dea,除此以外,与实施例7同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为176℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为160mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为174℃,耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1040mpa,是良好的,冲击后压缩强度为270mpa,微裂纹数为5个以下,是特别良好的。
[0162]
(实施例11~13)
[0163]
将各成分的含量和h/e如表1-2所示那样变更了,除此以外,与实施例2同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为170℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强
复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的温度。其中,实施例11、12为175℃以上且185℃以下,是特别优选的结果。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为200mpa
·
s以下,向增强纤维的含浸性也良好。其中,实施例11、12为180mpa
·
s以下,是特别优选的结果。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为170℃以上,耐热性也良好。其中,实施例11、12为175℃以上,是特别优选的结果。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,微裂纹数小于10个,力学特性、耐久性也良好。其中,实施例11中微裂纹数为5个以下,实施例12中湿热时0
°
压缩强度为1100mpa以上,冲击后压缩强度为270mpa以上,微裂纹数为5个以下,是特别优选的结果。
[0164]
(实施例14~16)
[0165]
将各成分的含量和h/e如表1-2、表1-3所示那样变更了,除此以外,与实施例7同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为170℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的温度。其中,实施例14、15为175℃以上且185℃以下,是特别优选的结果。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为200mpa
·
s以下,向增强纤维的含浸性也良好。其中,实施例14、15为180mpa
·
s以下,是特别优选的结果。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为170℃以上,耐热性也良好。其中,实施例14、15为175℃以上,是特别优选的结果。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,微裂纹数小于10个,力学特性、耐久性也良好。其中,实施例14中微裂纹数为5个以下,实施例15中湿热时0
°
压缩强度为1140mpa,冲击后压缩强度为275mpa,微裂纹数为5个以下,是特别优选的结果。
[0166]
(实施例17)
[0167]
如果表1-3所示那样,加入作为成分[a]的
“アラルダイト
(注册商标)”my721 65质量份、作为成分[b]的“epiclon(注册商标)”83020质量份、作为成分[c]的
“ロンザキュア
(注册商标)”m-cdea 61质量份、作为成分[d]的
“ロンザキュア
(注册商标)”m-dipa 26质量份、作为成分[f]的
“カネエース
(注册商标)”mx-416 20质量份,在80℃下搅拌1小时而调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为177℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为170mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为182℃,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1100mpa,冲击后压缩强度为280mpa,微裂纹数为5个以下,特别是力学特性、耐久性也良好。
[0168]
(实施例18、19)
[0169]
将各成分的含量和核壳橡胶粒子种类如表1-3所示那样变更了,除此以外,与实施
例17同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为175℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为200mpa
·
s以下,向增强纤维的含浸性也良好。其中,实施例19为170mpa
·
s,是特别优选的结果。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为175℃以上,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,力学特性也良好,微裂纹数为5个以下,特别是耐久性也良好。其中,实施例18中冲击后压缩强度为280mpa,是特别优选的结果。
[0170]
(实施例20)
[0171]
如表1-3所示那样,加入作为成分[a]的
“アラルダイト
(注册商标)”my721 65质量份、作为成分[b]的“epiclon(注册商标)”830 20质量份、作为成分[c]的
“ロンザキュア
(注册商标)”m-cdea 78质量份、作为成分[d]的
“ロンザキュア
(注册商标)”m-dipa 5质量份、作为成分[e]的
“ロンザキュア
(注册商标)”m-mipa 4质量份、作为成分[f]的
“カネエース
(注册商标)”mx-416 20质量份,在80℃下搅拌1小时而调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为184℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为120mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为180℃,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1120mpa,冲击后压缩强度为280mpa,微裂纹数为5个以下,特别是力学特性、耐久性也良好。
[0172]
(实施例21、22)
[0173]
将各成分的含量和核壳橡胶粒子种类如表1-4所示那样变更了,除此以外,与实施例20同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为175℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为180mpa
·
s以下,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为175℃以上,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,力学特性也良好,微裂纹数为5个以下,特别是耐久性也良好。其中,实施例21中湿热时0
°
压缩强度为1140mpa,冲击后压缩强度为275mpa,是特别优选的结果。
[0174]
(实施例23~26)
[0175]
变更了粘合剂种类,除此以外,与实施例2同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为175℃以上且185℃以下,在粘合剂的
熔点为165℃以上且180℃以下时粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都为180mpa
·
s以下,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果都为175℃以上,特别是耐热性也良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度为1000mpa以上,冲击后压缩强度为260mpa以上,微裂纹数小于10个,力学特性、耐久性也良好。其中,实施例21中微裂纹数为5个以下,实施例25中湿热时0
°
压缩强度为1100mpa,微裂纹数为5个以下,实施例26中微裂纹数为5个以下,是特别优选的结果。
[0176]
(比较例1、2)
[0177]
将各成分的含量如表2-1所示那样变更了,除此以外,与实施例1同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都为175℃以上且185℃以下,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果,比较例1为100mpa
·
s,特别是向增强纤维的含浸性也良好,但比较例2为210mpa
·
s,超过了200mpa
·
s,向增强纤维的含浸性差。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果,比较例1为168℃,低于170℃,耐热性差,但比较例2为196℃,特别是耐热性良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数。关于湿热时0
°
压缩强度,比较例1为990mpa,低于1000mpa,湿热时0
°
压缩强度差,但比较例2为1270mpa,是特别优选的结果。接下来,关于冲击后压缩强度,比较例1为285mpa,是特别优选的结果,但比较例2为245mpa,低于260mpa,耐冲击性差。进一步,关于微裂纹数,比较例1、2都为5个以下,特别是耐久性良好。
[0178]
(比较例3)
[0179]
代替成分[b],而使用了“epiclon(注册商标)”850和变更了各成分的含有比例,除此以外,与实施例2同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为177℃,粘合剂与树脂的界面粘接性良好并且保持粘合剂形状,因此为在纤维增强复合材料中可以均匀地确保能够进行充分的塑性变形的层间厚度的特别优选的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为180mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为187℃,特别是耐热性良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为980mpa,低于1000mpa,湿热时0
°
压缩强度差,但冲击后压缩强度为275mpa,特别是耐冲击性良好,微裂纹数为5个以下,特别是耐久性也良好。
[0180]
(比较例4)
[0181]
作为固化剂,仅使用了成分[c]和变更了各成分的含有比例,除此以外,与实施例2同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果为195℃,粘合剂在树脂中过度熔融,不保持粘合剂形状,因此为在纤维增强复合材料中不能
均匀地确保能够进行充分的塑性变形的层间厚度的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果为90mpa
·
s,特别是向增强纤维的含浸性也良好。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为178℃,特别是耐热性良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,湿热时0
°
压缩强度为1100mpa,是特别良好的结果,冲击后压缩强度为255mpa,低于260mpa,耐冲击性差,微裂纹数为5个以下,特别是耐久性良好。
[0182]
(比较例5~8)
[0183]
作为固化剂,仅使用了成分[d]或成分[e]或除成分[c]/成分[d]/成分[e]以外的固化剂一种和变更了各成分的含有比例,除此以外,与比较例4同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都低于170℃,在高于环氧树脂组合物的凝胶化温度的温度下将增强纤维基材连结的粘合剂熔化出,因此为界面粘接强度降低的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都大于1000mpa
·
s,超过了200mpa
·
s,向增强纤维的含浸性差。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为170℃以上,耐热性良好。其中,比较例5、6、7为175℃以上,是特别优选的结果。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度低于1000mpa,冲击后压缩强度低于260mpa,微裂纹数为10个以上,力学特性、耐久性不良。
[0184]
(比较例9~11)
[0185]
作为固化剂,代替成分[d],而使用了成分[e]或除成分[c]/成分[d]/成分[e]以外的固化剂一种和变更了各成分的含有比例,除此以外,与实施例2同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都低于170℃,在高于环氧树脂组合物的凝胶化温度的温度下将增强纤维基材连结的粘合剂熔化出,因此为界面粘接强度降低的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都超过了200mpa
·
s,向增强纤维的含浸性差。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为175℃以上,耐热性特别良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度低于1000mpa,冲击后压缩强度低于260mpa,微裂纹数为10个以上,力学特性、耐久性不良。
[0186]
(比较例12,13)
[0187]
作为固化剂,代替成分[d],而使用了除成分[c]/成分[d]/成分[e]以外的固化剂一种和变更了各成分的含有比例,除此以外,与实施例7同样地调制出环氧树脂组合物。如上述那样,测定了环氧树脂组合物的凝胶化温度,结果都低于170℃,在高于环氧树脂组合物的凝胶化温度的温度下将增强纤维基材连结的粘合剂熔化出,因此为界面粘接强度降低的温度。接下来,测定了120℃240分钟保持后的环氧树脂组合物的粘度,结果都超过了200mpa
·
s,向增强纤维的含浸性差。此外,通过上述方法而制作树脂固化物,测定了固化物的tg,结果为175℃以上,耐热性特别良好。进一步,使用该环氧树脂组合物而制作纤维增强复合材料,测定了湿热时0
°
压缩强度、冲击后压缩强度、微裂纹数,结果,都是湿热时0
°
压缩强度低于1000mpa,冲击后压缩强度低于260mpa,微裂纹数为10个以上,力学特性、耐久性不良。
[0188]
[表1-1]
[0189][0190]
[表1-2]
[0191][0192]
[表1-3]
[0193][0194]
[表1-4]
[0195][0196]
[表2-1]
[0197][0198]
[表2-2]
[0199][0200]
产业可利用性本发明的纤维增强复合材料用环氧树脂组合物由于向增强纤维基材的树脂注入中的树脂粘度长时间保持低粘度,因此工艺性良好,进一步,在有时加热固化时的升温速度慢的航空器主翼等巨大的结构材料中也可以将高水平的物性(耐热性、高压缩强度、耐冲击性、耐久性)赋予纤维增强复合材料。由此,特别是向航空器用途的纤维增强复合材料的应用进展,进一步可以期待由轻量化带来的燃耗提高、对地球变暖气体排出减少的贡献。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。