一种zn-mof-74金属有机骨架材料及制备和其在甲苯吸附中的应用
技术领域
1.本发明涉及有毒气体净化领域,具体涉及一种有毒气体甲苯吸附剂及其制备方法。
背景技术:
2.早在2017年,世界卫生组织国际癌症研究机构就将甲苯列为致癌物。对世界上很多城市空气中的平均浓度进行汇总,结果表明甲苯浓度通常为112.5-150μg/m3,这主要来自与汽油有关的排放,也来自于工业活动所造成的溶剂损失和排放。它不仅对环境有严重危害,对空气及水源造成污染;另外对人体健康也有严重影响,对皮肤、粘膜有刺激性,对中枢神经系统有麻醉作用,长期接触可发生神经衰弱综合征。
3.目前,现有的气体净化方式通常采用活性炭和天然沸石等物理吸附,但此类材料吸附量较小,消耗比较大,且吸附能力不强,使用一定的时间后会使吸附量变小,甚至失去吸附能力。另外,吸附时存在吸附的专一性问题,对混合气体,吸附性会减弱,存在被吸附物质的分子直径与孔径不匹配而导致的脱附现象。另外天然沸石孔道易堵塞,其表面硅氧结构具有极强的亲水性,结构外部阴离子易水解。
4.金属-有机骨架材料(metal-organic frameworks,mofs)作为21世纪的新型多功能材料。金属离子和有机配体之间是通过配位键连接在一起的,连接后形成了类蜂窝型规则有序的网络结构,且结构多样可调。由于配位键的作用力比较强,因此mofs的结构一般比其他弱相互作用力构筑的超分子体系的结构更稳定。mofs材料与传统无机沸石材料和多孔碳材料相比具有较高的孔隙率和较大的比表面积,可作为一种新型活性气体吸附材料,在环保领域具有较大的应用潜力。
技术实现要素:
5.在众多mofs材料中,mof-74以结构稳定性而著称。普通mofs材料结构所处环境发生变化时,它们的结构很容易就会被破坏,因此对气体的吸附量也会随之减少。但mof-74的稳定性优异,结构并不会随环境变化受到较大影响。其开放的金属位点与吸附分子之间形成有一定角度的配合物。通过物理化学吸附,达到较好的处理效果。
6.本发明所要解决的技术问题是针对上述现有问题,提出一种应用于甲苯吸附的zn-mof-74多孔金属有机骨架材料的制备方法:将硝酸锌和2,5-二羟基对苯二甲酸(dhta)溶解在n,n-二甲基甲酰胺dmf、无水乙醇和超纯水的混合溶剂中,将所得混合物溶液超声处理,随后转移至内衬是聚四氟乙烯的高压反应釜中保温一定时间,然后将其冷却至室温,离心收集产物。用dmf和甲醇洗涤几次,然后真空干燥得到zn-mof-74晶体。
7.本发明解决上述技术问题所采用的技术方案是:一种应用于甲苯吸附的zn-mof-74多孔金属有机骨架材料的制备方法,其特征在于包括有以下依次步骤:
8.1)将一定量的硝酸锌和2,5-二羟基对苯二甲酸(dhta)溶解在n,n-二甲基甲酰胺
dmf、无水乙醇和超纯水的混合溶剂中,然后将所得混合物溶液超声处理变成均相溶液;
9.2)将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入烘箱中保温一定时间,然后将其冷却至室温,离心收集产物;
10.3)用dmf和甲醇洗涤几次,然后真空干燥得到可以应用于甲苯吸附的zn-mof-74材料。
11.按上述方案,步骤1)所述的硝酸锌和2,5-二羟基对苯二甲酸的摩尔比为1-4。
12.按上述方案,步骤1)所述的n,n-二甲基甲酰胺dmf、无水乙醇和超纯水的体积比为1~3:1:1~0。
13.按上述方案,步骤1)所述混合物溶液超声处理的时间为5-20min。
14.按上述方案,步骤2)所述的保温温度为100-150℃,保温时间为12-24h。
15.按上述方案,步骤2)所述的离心转速为2000-8000r/min,离心时间为5-10min。
16.按上述方案,步骤3)所述的用dmf和甲醇洗涤次数为3-6次。
17.按上述方案,步骤3)所述的真空干燥的温度为60-80℃,干燥时间为6-12h。
18.本发明提出用水热法合成zn-mof-74,通过调节溶剂和溶质的比例,控制晶体的生长过程,最终得到zn-mof-74晶体。
19.zn-mof-74晶体的微观结构表征方法:用x射线衍射(xrd)光谱分析材料晶体结构,用扫描电镜(sem)观察材料形貌,利用热重分析(tga)分析样品的热稳定性,利用bet分析样品的孔径和比表面积,利用能谱仪(eds)分析材料的元素组成。
20.本发明的有益效果在于:本发明所述的zn-mof-74多孔金属有机骨架材料,采用简单的水热法合成,合成过程简单,操作方便。通过优化合成条件,合成了性能较好的zn-mof-74晶体,比表面积可达866m2/g。对甲苯这类危害环境及人体健康的有毒气体的吸附量可达308mg
·
g-1
。本发明具有设备要求低、无需昂贵的各种反应装置、易于大批量合成等优点,有望产生良好的社会和经济效益。
21.所得到的zn-mof-74晶体不仅具有很高的稳定性,六角边形的蜂窝网络拓扑结构为其提供了很大的比表面积和丰富的孔隙率。除对水蒸气和二氧化碳显示出较高的吸附性外,对甲苯这类有毒气体也表现出优异的吸附性能。zn-mof-74对甲苯的吸附作用主要来自于甲苯与金属位点之间的范德华力和静电力,甲苯主要吸附在金属位点上,并与金属原子形成有一定角度的配合物。
附图说明
22.图1为实施例1中zn-mof-74晶体的甲苯吸附曲线;
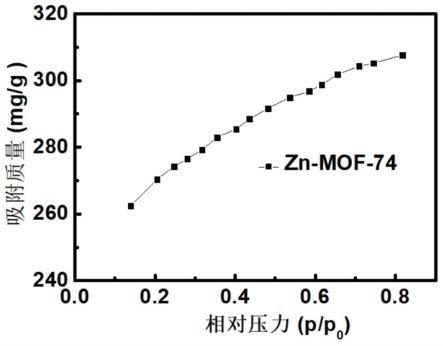
23.图2为实施例1中zn-mof-74晶体的xrd图谱;
24.图3为实施例1、2、3、4、5中zn-mof-74晶体的的sem图像;
25.其中:a)zn-mof-74(v
dmf
:v
无水乙醇
:v
超纯水
=1:1:1,n
zn(no3)2
·
6h2o
:n
dhta
=1:1);
26.b)zn-mof-74(v
dmf
:v
无水乙醇
:v
超纯水
=1:1:1,n
zn(no3)2
·
6h2o
:n
dhta
=2:1);
27.c)zn-mof-74(v
dmf
:v
无水乙醇
:v
超纯水
=1:1:1,n
zn(no3)2
·
6h2o
:n
dhta
=3:1);
28.d)zn-mof-74(v
dmf
:v
无水乙醇
:v
超纯水
=1:1:0,n
zn(no3)2
·
6h2o
:n
dhta
=3:1);
29.e)zn-mof-74(v
dmf
:v
无水乙醇
:v
超纯水
=3:1:1,n
zn(no3)2
·
6h2o
:n
dhta
=3:1).
30.图4为实施例1中zn-mof-74晶体的tg曲线;
31.图5为实施例1中zn-mof-74晶体的n2吸附-脱附曲线;
32.表1为实施例1中zn-mof-74晶体的eds元素分析;
33.表2为实施例1中zn-mof-74晶体的孔结构参数。
具体实施方式
34.下面结合实施例对本发明做进一步详细的说明,但是此说明不会构成对本发明的限制。
35.实施例1:
36.将3.2145g的六水合硝酸锌zn(no3)2·
6h2o和0.7369g的2,5-二羟基对苯二甲酸(dhta)混合后溶解在10ml的n,n-二甲基甲酰胺dmf、10ml无水乙醇和10ml超纯水的混合溶剂中,然后将所得混合物溶液超声处理约10分钟变成均相溶液,随后将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入100℃的烘箱中24小时。之后将其冷却至室温,通过5000r/min离心5min收集淡黄色产物,,最后将其依次用dmf和甲醇各洗涤2次,然后60℃真空干燥12h得到zn-mof-74黄色晶体。
37.图1是zn-mof-74甲苯吸附曲线。zn-mof-74前处理条件为60℃真空干燥6小时,测试温度25℃,甲苯蒸汽在相对压力p/p0=0-1压力整个范围内(p0是标准大气压),zn-mof-74材料对甲苯的吸附量逐渐增加,饱和吸附量可以达到308mg
·
g-1
,zn-mof-74对甲苯的吸附作用主要来自于甲苯与金属位点之间的范德华力和静电力,甲苯主要吸附在金属位点上,并与金属原子形成有一定角度的配合物。
38.图2是zn-mof-74晶体的xrd图谱。zn-mof-7晶体在2θ=6.737
°
位置处和2θ=11.698
°
位置处有明显的衍射峰,这与之前报道的mof-74的衍射峰的峰形和位置一样,图中虽然有少量杂峰,但是并不会对m-mof-74的结构造成影响。
39.图3是zn-mof-74晶体的sem图像。分别为不同溶质比(溶剂均为体积比dmf:无水乙醇:超纯水=1:1:1)、不同溶剂比(溶质均为摩尔比zn(no3)2·
6h2o:dhta=3:1)的zn-mof-74的扫描电镜图。从图中可以看出,溶质比为3:1的晶体形貌最好,一定程度上,晶体形貌的好坏会直接影响晶体性能,说明zn(no3)
·
6h2o的过量能有利产物zn-mof-74的结晶,同时也可以说明配体dhta过量不利于晶体结晶。从图中可以看出,不同溶剂比条件下制得的zn-mof-74材料形貌相差不大,这说明dmf虽然有助于dhta的溶解,但是dmf过量并不有助于晶体的生长。为此,在合成过程中,我们要选择适宜的溶质比,以及适宜的dmf溶剂。
40.图4是zn-mof-74晶体的tg曲线,在n2气氛下,加热速率10
°
min-1
,范围为40-800℃。zn-mof-74在100℃前失重较大,这是由于孔道中水分子的蒸发引起的;在100-300℃间质量基本无变化;zn-mof-74的质量进一步减小发生在300℃之后,这可归因于配体与金属的配位键断裂,zn-mof-74结构发生坍塌。
41.图5是zn-mof-74晶体的n2吸附-脱附曲线,脱气温度100℃,脱气时间120min,环境温度20℃,吸附质n2。在低压区域(p/p0=0-0.1),对氮气的吸附很快的便达到了饱和量200cm3/g;在中压区域(p/p0=大于0.1至0.8),吸附氮气的量随着压力的增大变化程度几乎可以忽略;但当到高压区域时(p/p0=大于0.8至1),氮气的吸附量随着压力迅速增大。从图中可以看出吸附曲线与脱附曲线基本吻合的很好,但是在高压区域氮气的脱附曲线相比吸附曲线仍存在轻微的延后,这可能是由于zn-mof-74晶体材料中存在的一些大孔造成的。
42.表1为zn-mof-74晶体的eds元素分析,zn元素的含量仅占大约15%,碳元素含量最多,大约占到了63%,氧元素约占21%,碳、锌、氧含量比约为12:3:4。
43.表2为zn-mof-74晶体的孔结构参数,其中zn-mof-74晶体的中孔直径约为11nm、bet单点法比表面约为866m2/g、langmuir法比表面积约为942m2/g,延长真空干燥时间来清除孔隙中的客体分子(水分子),比表面积还会有略微增加。
44.表1zn-mof-74中各元素含量一览表
[0045][0046][0047]
表2zn-mof-74晶体的孔结构参数
[0048][0049]
中孔孔径范围为2-50nm,微孔孔径范围为小于2nm;
[0050]
实施例2:
[0051]
为了检验溶质比对zn-mof-74晶体材料的影响,改变溶质比即六水合硝酸锌zn(no3)2·
6h2o和2,5-二羟基对苯二甲酸的摩尔比为2:1,其反应过程如下:将2.1430g的六水合硝酸锌zn(no3)2·
6h2o和0.7369g的2,5-二羟基对苯二甲酸(dhta)混合后溶解在10ml的n,n-二甲基甲酰胺dmf、10ml无水乙醇和10ml超纯水的混合溶剂中,然后将所得混合物溶液超声处理约10分钟变成均相溶液,随后将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入100℃的烘箱中24小时。之后将其冷却至室温,通过5000r/min离心5min收集淡黄色产物,,最后将其用dmf和甲醇依次各洗涤2次,然后60℃真空干燥12h,经xrd验证得到zn-mof-74黄色晶体。
[0052]
实施例3:
[0053]
为了检验溶质比对zn-mof-74晶体材料的影响,改变溶质比即六水合硝酸锌zn
(no3)2·
6h2o和2,5-二羟基对苯二甲酸的摩尔比为1:1,其反应过程如下:将1.0715g的六水合硝酸锌zn(no3)2·
6h2o和0.7369g的2,5-二羟基对苯二甲酸(dhta)混合后溶解在10ml的n,n-二甲基甲酰胺dmf、10ml无水乙醇和10ml超纯水的混合溶剂中,然后将所得混合物溶液超声处理约10分钟变成均相溶液,随后将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入100℃的烘箱中24小时。之后将其冷却至室温,通过5000r/min离心5min收集淡黄色产物,,最后将其用dmf和甲醇依次各洗涤2次,然后60℃真空干燥12h,经xrd验证得到zn-mof-74黄色晶体。
[0054]
实施例4:
[0055]
为了检验溶剂对zn-mof-74晶体材料的影响,改变溶剂比即n,n-二甲基甲酰胺dmf、无水乙醇和超纯水的体积比为1:1:0,其反应过程如下:将3.2145g的六水合硝酸锌zn(no3)2·
6h2o和0.7369g的2,5-二羟基对苯二甲酸(dhta)混合后溶解在15ml的n,n-二甲基甲酰胺dmf、15ml无水乙醇的混合溶剂中,然后将所得混合物溶液超声处理约10分钟变成均相溶液,随后将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入100℃的烘箱中24小时。之后将其冷却至室温,通过5000r/min离心5min收集淡黄色产物,,最后将其用dmf和甲醇依次各洗涤2次,然后60℃真空干燥12h,经xrd验证得到zn-mof-74黄色晶体。
[0056]
实施例5:
[0057]
为了检验溶剂对zn-mof-74晶体材料的影响,改变溶剂比即n,n-二甲基甲酰胺dmf、无水乙醇和超纯水的体积比为3:1:1,其反应过程如下:将3.2145g的六水合硝酸锌zn(no3)2·
6h2o和0.7369g的2,5-二羟基对苯二甲酸(dhta)混合后溶解在18ml的n,n-二甲基甲酰胺dmf、6ml无水乙醇和6ml超纯水的混合溶剂中,然后将所得混合物溶液超声处理约10分钟变成均相溶液,随后将溶液转移至内衬是聚四氟乙烯的高压反应釜中,并放入100℃的烘箱中24小时。之后将其冷却至室温,通过5000r/min离心5min收集淡黄色产物,,最后将其用dmf和甲醇依次各洗涤4次,然后60℃真空干燥12h,经xrd验证得到zn-mof-74黄色晶体。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。