1.本发明属于催化剂领域,特别涉及一种负载型钒铬双中心催化剂及其制备方法与应用。
背景技术:
2.聚乙烯具备优良的力学性能和加工性能而被广泛应用于国民生活的各个领域,比如食品包装薄膜、吹塑容器、农用薄膜、给排水管道、汽车零部件、电子器件等。聚乙烯的消费量也在逐年递增,目前全球年产量已突破1亿吨,因此发展聚乙烯合成用催化新技术具有重要意义。
3.目前,工业上广泛应用的合成聚乙烯的催化剂主要包含如下三大类:ziegler~natta钛系催化剂、铬基催化剂(主要包括无机铬phillip催化剂和有机铬s~2催化剂)以及茂金属催化剂。上述催化剂聚乙烯产品的普遍特点为分子量呈单峰分布,且其乙烯/α-烯烃共聚物中短支链分布一般倾向于集中分布与低分子量部分(使用茂金属催化剂得到的产物则基本沿分子量均匀分布)。但是高分子物理研究结果表明,相比于单峰聚乙烯,分子量呈宽、双峰分布的聚乙烯产品其力学性能更优,同时还兼具优良的加工性能。此外,引入少量短支链于聚乙烯的高分子量组分也可明显提升其产品的长期力学性能,如抗慢速裂纹增长(krishnaswamy,r.k.;yang,q.;fernandez-ballester,l.;kornfield,j.a.effect of the distribution of short-chain branches on crystallization kinetics and mechanical properties of high-density polyethylene.macromolecules.2008,41,1693-1704.)。为了合成性能优异的短支链分布于高分子量组分的双峰聚乙烯,目前工业上普遍采用两个或多个反应釜串联操作,并通过调控各个反应釜链转移剂和共聚单体浓度的方法实现。但是显然该方法的设备投资和操作费用也随之有所增加。
4.与此同时,工业界和学术界也正在积极探索新的更加绿色和低成本的生产工艺。其中报道的较多的是开发复合型双中心催化剂,即将两种催化剂共混或者共负载于某一载体,其研究的出发点在于使该类催化剂能够同时具备两种活性中心的特性,甚至利用两种活性中心间的协同效应,表现出更为优异的催化特性。双金属中心聚乙烯催化剂(cn103626899a和cn104448067a),其双金属中心间在大多数条件下表现出优良的催化协同效应,使催化剂的聚合活性、共聚性能等方面在原来单金属催化剂的基础上有明显改善,且合成聚合物的分子量呈宽峰或者双峰分布,共聚单体也集中分布于高分子量组分,因此该系列催化剂在单釜法合成高性能双峰聚乙烯方面拥有巨大优势,有可能部分取代工业上常用的合成高性能双峰聚乙烯料的双釜串联工艺,因为相对于后者,利用双中心催化技术于单一反应釜中合成高性能双峰聚乙烯的设备投资更低,操作难度更小,能耗也更低。
技术实现要素:
5.本发明的主要目的在于提供一种负载型双中心催化剂及其制备方法与应用,以克服现有技术双中心催化剂聚合活性较低,共聚单体插入率较低的缺陷。
6.为了达到上述目的,本发明提供了一种负载型双中心催化剂的制备方法,该负载型双中心催化剂包括载体和活性组分,该制备方法包括如下步骤:
7.步骤1,将载体浸渍于改性剂溶液中进行浸渍反应,所述改性剂溶液包括改性剂,所述改性剂为有机铝化合物;
8.步骤2,将改性后的载体负载活性组分的前驱体,所述活性组分的前驱体包括有机铬源和无机钒源,得到负载型双中心催化剂。
9.本发明所述的负载型双中心催化剂的制备方法,其中,所述载体在浸渍改性剂溶液前还包括焙烧操作,以脱除载体表面的水和部分羟基。
10.本发明所述的负载型双中心催化剂的制备方法,其中,改性后的载体负载活性组分的前驱体的方式为分步负载有机铬源和无机钒源,或者为同时负载有机铬源和无机钒源。
11.本发明所述的负载型双中心催化剂的制备方法,其中,所述焙烧操作的温度为200~900℃,焙烧过程的升温速率为0.1~10℃/min,焙烧时间为1~10h;步骤2的负载方法为浸渍。
12.本发明所述的负载型双中心催化剂的制备方法,其中,所述载体为氧化硅、氧化铝、硅铝酸盐、无机粘土、氧化钛、氧化锆、氧化镁、氧化铁、氧化锡、氧化锌中的至少一种;所述载体的比表面积为100~1000m2/g,孔体积为0.3~5.0cm3/g,平均孔径为5~50nm。
13.本发明所述的负载型双中心催化剂的制备方法,其中,所述有机铝化合物为三烷基铝alr3、二烷基烷氧基铝alr2or、二烷基卤化铝alr2x、铝氧烷、乙基倍半铝氯化物中的至少一种,其中r为具有1-12个碳原子的烷基,x为卤素。
14.本发明所述的负载型双中心催化剂的制备方法,其中,所述有机铬源含有环戊二烯基配体、茚基配体、芴基配体中的至少一种;所述无机钒源为含钒卤化物。
15.本发明所述的负载型双中心催化剂的制备方法,其中,所述有机铬源的结构简式为:crcp1cp2,其中cp1与cp2独立为环戊二烯基、茚基和芴基中的一种,且cp1与cp2相同或不相同;所述钒源选自三氟化钒、三氯化钒、三溴化钒、四氟化钒、四氯化钒、二氟氧钒、二氯氧钒、二溴氧钒、三氟氧钒、三氯氧钒中的至少一种。
16.为了达到上述目的,本发明还提供了一种负载型双中心催化剂,由上述的负载型双中心催化剂的制备方法制得,以该负载型双中心催化剂的总质量为基准,该负载型双中心催化剂中,活性组分以金属计,cr的含量为0.1wt%~8wt%,v的含量为0.1wt%~10wt%,改性剂以金属计的含量为0.1~10wt%。
17.为了达到上述目的,本发明更提供了一种烯烃聚合方法,使用上述的负载型双中心催化剂。
18.本发明所述的烯烃聚合方法,其中,包括所述负载型双中心催化剂的活化步骤,活化试剂为烷基铝化合物。
19.本发明的有益效果:
20.1、本发明双活性中心催化剂中两种活性中心能够较好地表现各自的催化性能,而且还表现出良好的催化协同效应,同时对催化剂载体的改性处理能够进一步改善催化剂的催化活性;
21.2、本发明双金属中心催化剂相比于传统的单、双中心催化剂,在用于烯烃聚合时,
可以使聚合物分子量分布更宽,呈双峰分布,包含由有机铬活性中心合成的较低分子量乙烯均聚物a,由有机钒活性中心合成的较高分子量乙烯共聚物b,且a与b两种组分的相对含量可主要地通过调整催化剂中两种活性中心的相对含量进行调节;
22.3、本发明双金属中心催化剂的两种活性中心对活化试剂烷基铝化合物有很强的敏感性,即含氯活化试剂对钒组分有很强的活化作用,但是对有机铬组分的活化作用却很弱,利用两种铬、钒组分对助催化剂的差异化反应,可通过调节活化剂组分较灵敏地调控聚合物中a与b组分的相对含量;
23.4、本发明双金属中心催化剂载体经过改性剂处理,还可提高该催化剂的共聚效率,即在相同的共聚单体加入量下,载体经过改性剂处理的双金属中心催化剂其共聚单体插入率更高;
24.因此,本发明负载型双活性中心催化剂可在单一反应器中生产分子量分布较宽,甚至呈双峰分布的乙烯均聚物和乙烯/α-烯烃共聚物,其含有较低分子量的乙烯均聚物和较高分子量乙烯与α-烯烃的共聚物。该催化剂还具有较高的乙烯均聚和乙烯与α-烯烃共聚反应活性。
具体实施方式
25.以下对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和过程,但本发明的保护范围不限于下述的实施例,下列实施例中未注明具体条件的实验方法,通常按照常规条件。
26.本发明提供了一种负载型双中心催化剂的制备方法,该负载型双中心催化剂包括载体和活性组分,该制备方法包括如下步骤:
27.步骤1,将载体浸渍于改性剂溶液中进行浸渍反应,所述改性剂溶液包括改性剂,所述改性剂为有机铝化合物;
28.步骤2,将改性后的载体负载活性组分的前驱体,所述活性组分的前驱体包括有机铬源和无机钒源,得到负载型双中心催化剂。
29.在一实施方式中,载体在浸渍改性剂溶液前还包括焙烧操作,以脱除载体表面的水和部分羟基。
30.焙烧操作在流化状态或非流化状态下均可进行,但优选在流化状态下进行。在一实施方式中,焙烧操作是在氮气或氩气等惰性气氛中进行,以防止载体中残余氧气在后续过程中与铬源或钒源反应。焙烧的温度可以为200~900℃,优选为300~600℃,焙烧过程的升温速率为0.1~10℃/min,优选3~5℃/min,焙烧时间可以为1~10h,优选为2~4h。载体表面的羟基密度对活性组分具有双重影响,一方面过高密度的羟基对活性中心有毒化作用,而过低密度的羟基将不利于铬源与钒源的负载,因此本发明通过对载体进行焙烧,脱除部分羟基,使催化剂载体表面具有合适的羟基密度。
31.在一实施方式中,本发明载体为多孔无机载体,可以为氧化硅、氧化铝、硅铝酸盐、无机粘土、氧化钛、氧化锆、氧化镁、氧化铁、氧化锡、氧化锌中的至少一种;其中,无机粘土优选为蒙脱石,氧化硅优选为无定型多孔硅胶。在另一实施方式中,本发明载体的比表面积为100~1000m2/g,孔体积为0.3~5.0cm3/g,平均孔径在5~50nm。
32.在一实施方式中,用于载体改性的改性剂为有机铝化合物,包括烷基铝及其衍生
物,如三烷基铝alr3、二烷基烷氧基铝alr2or、二烷基卤化铝alr2x、铝氧烷、乙基倍半铝氯化物等,其中r是烷基,例如具有1-12个碳原子的烷基,更例如是甲基、乙基、正丙基、异丙基、正丁基、异丁基、正戊基、正己基、正庚基、正辛基、正壬基、正十二烷基等,x是卤素,例如氟、氯、溴和碘,优选氯。其中,铝氧烷为烷基铝与水的反应物。本发明中改性剂可以单独使用或两种或两种以上组合使用。作为具体例子,改性剂可以为三乙基铝、三异丁基铝、二乙基乙氧基铝、一氯二乙基铝和甲基铝氧烷中的至少一种。
33.在一实施方式中,载体进行改性处理的步骤为:将焙烧处理后的载体与改性剂有机铝化合物置于有机溶剂中进行浸渍反应,得到改性载体。本发明上述浸渍过程中的有机溶剂为惰性有机溶剂,通过浸渍搅拌的方法使有机铝化合物进入多孔无机载体孔道,并吸附于其表面。详细而言,有机铝化合物会进入载体孔道内会与羟基反应,进而锚定在载体表面,也会物理吸附于载体孔道内表面。不仅可以脱除硅胶载体表面的部分物理吸附水与羟基,还有利于催化剂活性组分与有机铝化合物的相互作用。
34.在另一实施方式中,惰性有机溶剂可以为正己烷、正庚烷、正辛烷、苯、甲苯、二甲苯等中的至少一种。浸渍的温度为20~180℃,优选为45~120℃,浸渍时间为1~24h,优选为4~12h;浸渍结束后通过高温将体系中的溶剂蒸发脱除以达到干燥的目的,所述的干燥是指在60~150℃干燥,优选在80~130℃干燥,所述的干燥时间为2~24h,优选为6~16h。
35.在又一实施方式中,改性剂以al计占催化剂总重量的0.1~10wt%。
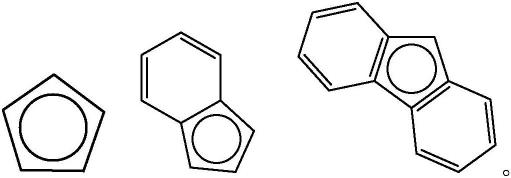
36.在一实施方式中,本发明有机铬源含有环戊二烯基配体、茚基配体、芴基配体中的至少一种;无机钒源为含钒卤化物。在另一实施方式中,本发明有机铬源的结构简式为:crcp1cp2,其中cp1与cp2独立为环戊二烯基、茚基和芴基中的一种,且cp1与cp2相同或不相同;具体地,cp1与cp2的结构为如下结构中的一种:
[0037][0038]
在另一实施方式中,本发明钒源选自三氟化钒、三氯化钒、三溴化钒、四氟化钒、四氯化钒、二氟氧钒、二氯氧钒、二溴氧钒、三氟氧钒、三氯氧钒中的至少一种。
[0039]
在一实施方式中,本发明改性后的载体负载活性组分的前驱体的方法为浸渍。本发明并不特别限定浸渍铬活性组分和钒活性组分的先后顺序,可以同时浸渍,也可以分步浸渍。
[0040]
在另一实施方式中,本发明载体浸渍活性组分为:将活性组分前驱体溶于惰性有机溶剂中,通过浸渍搅拌的方法使钒源和铬源进入多孔无机载体孔道,并吸附于其表面。其中惰性有机溶剂可以为正己烷、正庚烷、正辛烷、苯、甲苯、二甲苯等中的至少一种。铬源和钒源的用量满足:无机钒活性组分以v计占催化剂总重量的0.1~10wt%;有机铬活性组分以cr计占催化剂总重量的0.1~8wt%。其中,浸渍的温度可以为20~180℃,优选为45~120℃,浸渍的时间可以为1~24h,优选为4~12h;浸渍结束后通过高温将体系中的溶剂蒸发脱除以达到干燥的目的,所述的干燥是指在60~150℃干燥,优选在80~130℃干燥,所述的干燥时间为2~24h,优选为6~16h。
[0041]
本发明是利用焙烧活化的载体浸渍活性组分的前驱体得到催化剂,如此即可实现活性金属负载于载体上,因此所得到的催化剂不需要传统的焙烧步骤,可以防止活性金属在焙烧过程中的聚集现象。
[0042]
作为一具体实施方式,本发明提供了一种负载型双中心催化剂的制备方法,包括以下步骤:
[0043]
(1.1)高温焙烧载体以脱除载体表面的物理水以及部分羟基;
[0044]
(1.2)将(1.1)处理后的载体与改性剂(有机铝化合物)置于有机溶剂中反应,得到改性载体;
[0045]
(1.3)将(1.2)中所得改性载体与有机铬源共浸渍于有机溶剂中进行反应,形成载体表面负载有有机铬的催化剂前驱体;
[0046]
(1.4)将(1.3)中所得产物与钒源共浸渍于有机溶剂中进行反应,形成载体表面负载有有机铬和无机钒的催化剂;
[0047]
上述步骤(1.3)和(1.4)进行的顺序无特定要求,既可以先进行步骤(1.3)再进行步骤(1.4),也可以先进行步骤(1.4)再进行步骤(1.3),或者(1.3)与(1.4)同时进行,即有机铬源、无机钒源与改性载体在有机溶剂下共浸渍。
[0048]
本发明所制备的负载型双中心催化剂,以该负载型双中心催化剂的总质量为基准,该负载型双中心催化剂中,活性组分以金属计,cr的含量为0.1wt%~8wt%,v的含量为0.1wt%~10wt%,改性剂以金属计的含量为0.1~10wt%。
[0049]
本发明负载型双中心催化剂可用于乙烯均聚以及乙烯与α-烯烃的共聚,聚合单体可以为乙烯或常见α-烯烃,常见α-烯烃包括丙烯、1-丁烯、1-己烯、1-辛烯,其合成的聚合物分子量呈宽、双峰分布,包含较低分子量乙烯均聚物和较高分子量乙烯共聚物。
[0050]
本发明负载型双中心催化剂在催化烯烃聚合时,或者在用于烯烃聚合前,还包括催化剂的活化步骤,活化试剂为烷基铝化合物。
[0051]
在一实施方式中,烷基铝化合物对负载型双活性中心催化剂进行预还原活化处理是在惰性气氛下进行,所述的烷基铝化合物的摩尔量和负载型双活性中心催化剂中铬元素的总摩尔量的比值为0~1000,优选为0-100,更优选0-50;所述的预还原活化处理温度在室温~100℃之间,优选室温~60℃之间,还原活化处理时间0.5~20小时,优选0.5~10小时,还原活化处理方式可以为将催化剂和活化剂置于有机溶剂中进行浸渍,可以进行搅拌,优选连续搅拌,处理完毕后再在60~120℃之间干燥2~8小时,干燥在氮气或惰性气体气氛下进行,例如在氮气、氦气、氩气等气氛下进行,优选在氮气气氛下进行,该干燥过程也可在真空条件下进行,得到的经过预还原活化的负载型双活性中心催化剂在惰性气体气氛下保存待用。
[0052]
负载型双活性中心催化剂的活化步骤中,负载型双活性中心催化剂与烷基铝化合物反应原位形成目标催化剂是指在溶剂介质中进行原位还原活化处理,所述的溶剂介质可以选自异戊烷、正戊烷、正己烷、异己烷、正庚烷、正辛烷、甲苯、二甲苯,优选异戊烷、正己烷、异己烷和正庚烷中的至少一种;所述的烷基铝化合物的摩尔量和负载型双中心催化剂中铬元素的摩尔量的比值为0~1000,优选为0-100,更优选0-50;所述的原位还原活化处理的温度为20~120℃,原位还原活化处理的时间为0.5~5h。
[0053]
在实施方式i中,本发明负载型双活性中心催化剂的制备方法,包含如下步骤:
[0054]
(i)将多孔无机载体在200~900℃下焙烧活化;
[0055]
(ii)将经焙烧后的多孔无机载体浸渍于含烷基铝的溶液中反应一段时间,然后干燥备用;
[0056]
(iii)将(ii)中所得产物浸渍于含钒源的溶液反应一段时间,然后干燥保存;
[0057]
(iv)将(iii)中所得产物浸渍于含有机铬源的溶液,然后干燥,得到所述催化剂保存备用;
[0058]
根据一个优选的负载型双活性中心催化剂的制备方法,包含步骤:
[0059]
a)将多孔无机载体置于惰性气体中进行高温焙烧,焙烧温度在200~900℃,优选300~600℃,焙烧时间为1~10h,优选2~4h,然后自然冷却至室温后保存备用。
[0060]
b)将焙烧后的无机载体浸渍于含烷基铝的有机溶剂中,反应时间为1~12h,优选1~4h,反应温度为室温至180℃,优选45~120℃,然后升温至60~210℃下干燥,优选80~150℃,干燥时间为2~12h,优选2~4h,干燥后的产物可转移至惰性气体保护下保存待用。
[0061]
c)将b)中产物浸渍于含钒源的溶液进行反应负载,浸渍时间为1~24h,优选4~12h,浸渍温度为20~180℃,优选45~120℃,然后在60~150℃之间干燥,优选80~130℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,然后将干燥后混合物置于惰性气体保护下保存备用。
[0062]
d)将上述产物置于含有机铬源的溶液中进行浸渍反应负载,浸渍反应时间为1~24h,优选4~8h,浸渍温度为20~120℃,优选45~80℃,然后在60~150℃之间干燥,优选80~100℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,干燥结束后即得所述催化剂。
[0063]
作为一个实例,制备本发明催化剂的具体操作包括:
[0064]
将sio2载体置于流化床内进行高温焙烧,焙烧过程在氮气氛围下进行,焙烧温度为600℃。焙烧达到指定时间后(如2~4h),停止加热,并使干燥和部分脱羟基的sio2自然冷却至室温,然后收集保存在有惰性气体(如氮气)保护的环境下。将上述焙烧后的载体与一定浓度的三乙基铝甲苯溶液共混反应,三乙基铝以铝计的加入量相对催化剂总重量为1wt%,反应在80℃下进行,并施以连续搅拌,反应约2h后,升温至130℃干燥4~6h,干燥过程通以氮气,以加速干燥。干燥好的改性载体置于惰性气体保护下保存待用。将上述产物浸渍在含一定浓度vocl3正己烷溶液中,钒的加入量相对于催化剂总重量符合本发明的要求(例如v的加入量为0.2~3wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,用正己烷在一定温度下(如室温~60℃)洗涤催化剂,再于一定温度下(如60~120℃)干燥4~6小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。将上述负载了一定钒催化剂前驱体的产物浸渍在含一定浓度双环戊二烯基铬的正己烷溶液中,铬的加入量相对于催化剂总重量符合本发明的要求(例如cr的加入量为0.2~2wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,再于一定温度下(如60~120℃)干燥4~6小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。
[0065]
在实施方式ii中,本发明提供负载型双中心催化剂的制备方法,包含如下步骤:
[0066]
(i)将多孔无机载体在200~900℃下焙烧活化;
[0067]
(ii)将经焙烧后的多孔无机载体浸渍于含烷基铝的溶液中反应一段时间,然后干
燥备用;
[0068]
(iii)将(ii)中所得产物浸渍于含有机铬源的溶液反应一段时间,然后干燥保存;
[0069]
(iv)将(iii)中所得产物浸渍于含钒源的溶液,然后干燥,得到所述催化剂前驱体保存备用;
[0070]
根据一个优选的负载型双活性中心乙烯聚合催化剂的制备方法,包含步骤:
[0071]
a)将多孔无机载体置于惰性气体中进行高温焙烧,焙烧温度在200~900℃,优选300~600℃,焙烧时间为1~10h,优选2~4h,然后自然冷却至室温后保存备用。
[0072]
b)将焙烧后的无机载体浸渍于含烷基铝的有机溶剂中,反应时间为1~12h,优选1~4h,反应温度为室温至180℃,优选45~120℃,然后升温至60~210℃下干燥,优选80~150℃,干燥时间为2~12h,优选2~4h,干燥后的产物可转移至惰性气体保护下保存待用。
[0073]
c)将上述产物置于含有机铬源的溶液中进行浸渍反应负载,浸渍反应时间为1~24h,优选4~8h,浸渍温度为20~120℃,优选45~80℃,然后在60~150℃之间干燥,优选80~100℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,干燥结束后即得所述催化剂前驱体。
[0074]
d)将上述产物浸渍于含钒源的溶液进行反应负载,浸渍时间为1~24h,优选4~12h,浸渍温度为20~180℃,优选45~120℃,然后在60~150℃之间干燥,优选80~130℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,然后将干燥后混合物置于惰性气体保护下保存备用。
[0075]
作为一个实例,制备本发明催化剂的具体操作包括:
[0076]
将sio2/tio2(含5wt%ti)载体置于流化床内进行高温焙烧,焙烧过程在氮气氛围下进行,焙烧温度为600℃。焙烧达到指定时间后(如2~4h),停止加热,并使干燥和部分脱羟基的sio2/tio2自然冷却至室温,然后收集保存在有惰性气体(如氮气)保护的环境下。将上述焙烧后的载体与一定浓度的三异丁基铝正己烷溶液共混反应,三异丁基铝以铝计的加入量相对催化剂总重量为1wt%,反应在45℃下进行,并施以连续搅拌,反应约2h后,升温至80℃干燥4~6h,干燥过程通以氮气,以加速干燥。干燥好的改性载体置于惰性气体保护下保存待用。将上述改性载体浸渍在含一定浓度双茚基铬的正己烷溶液中,铬的加入量相对于催化剂总重量符合本发明的要求(例如cr的加入量为0.2~1wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,再于一定温度下(如60~120℃)干燥4~8小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。将上述负载了一定有机铬催化剂前驱体的产物浸渍在含一定浓度vocl3正己烷溶液中,钒的加入量相对于催化剂总重量符合本发明的要求(例如v的加入量为0.2~2wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,用正己烷在一定温度下(如室温~60℃)洗涤催化剂,再于一定温度下(如60~120℃)干燥4~6小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。
[0077]
在实施方式iⅱ中,本发明提供负载型双中心催化剂的制备方法,包含如下步骤:
[0078]
(i)将多孔无机载体在200~900℃下焙烧活化;
[0079]
(ii)将经焙烧后的多孔无机载体浸渍于含烷基铝的溶液中反应一段时间,然后干燥备用;
[0080]
(iii)将(ii)中所得产物浸渍于含有机铬源和钒源的溶液反应一段时间,然后干燥保存;
[0081]
根据一个优选的负载型双活性中心乙烯聚合催化剂的制备方法,包含步骤:
[0082]
a)将多孔无机载体置于惰性气体中进行高温焙烧,焙烧温度在200~900℃,优选300~600℃,焙烧时间为1~10h,优选2~4h,然后自然冷却至室温后保存备用。
[0083]
b)将焙烧后的无机载体浸渍于含烷基铝的有机溶剂中,反应时间为1~12h,优选1~4h,反应温度为室温至180℃,优选45~120℃,然后升温至60~210℃下干燥,优选80~150℃,干燥时间为2~12h,优选2~4h,干燥后的产物可转移至惰性气体保护下保存待用。
[0084]
c)将上述产物置于含有机铬源和钒源的溶液中进行浸渍反应负载,浸渍反应时间为2~24h,优选4~8h,浸渍温度为20~120℃,优选45~80℃,然后在60~150℃之间干燥,优选80~120℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,干燥结束后即得所述催化剂前驱体,然后将干燥产物置于惰性气体保护下保存备用。
[0085]
作为一个实例,制备本发明催化剂的具体操作包括:
[0086]
将sio2载体置于流化床内进行高温焙烧,焙烧过程在氮气氛围下进行,焙烧温度为500℃。焙烧达到指定时间后(如2~4h),停止加热,并使干燥和部分脱羟基的sio2自然冷却至室温,然后收集保存在有惰性气体(如氮气)保护的环境下。将上述焙烧后的载体与一定浓度的三乙基铝甲苯溶液共混反应,三乙基铝以铝计的加入量相对催化剂总重量为1wt%,反应在80℃下进行,并施以连续搅拌,反应约2h后,升温至130℃干燥4~6h,干燥过程通以氮气,以加速干燥。干燥好的改性载体置于惰性气体保护下保存待用。
[0087]
将上述产物浸渍在含一定浓度双环戊二烯基铬和vocl3的正己烷溶液中,铬和钒的加入量相对于催化剂总重量符合本发明的要求(例如cr的加入量为0.2~2wt%,v的加入量为0.2~1.5wt%);在一定温度下(如45-60℃)连续搅拌反应一定时间(例如4~8小时)后,用正己烷在一定温度下(如室温~60℃)洗涤催化剂,再于一定温度下(如60~120℃)干燥4~8小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。
[0088]
在实施方式iv中,本发明提供负载型双中心催化剂的制备方法,包含如下步骤:
[0089]
(i)将多孔无机载体在200~900℃下焙烧活化;
[0090]
(ii)将经焙烧后的多孔无机载体浸渍于含烷基铝的溶液中反应一段时间,然后干燥备用;
[0091]
(iii)将(ii)中所得产物浸渍于含钒源的溶液反应一段时间,然后干燥保存;
[0092]
(iv)将(iii)中所得产物浸渍于含有机铬源的溶液,然后干燥,得到所述催化剂保存备用;
[0093]
(v)将(iv)中所得催化剂浸渍于有机金属助催化剂溶液中进行预还原活化,得到具备催化活性的催化剂。
[0094]
根据一个优选的制备负载型双活性中心乙烯聚合催化剂的方法包含步骤:
[0095]
a)将多孔无机载体置于惰性气体中进行高温焙烧,焙烧温度在200~900℃,优选300~600℃,焙烧时间为1~10h,优选2~4h,然后自然冷却至室温后保存备用。
[0096]
b)将焙烧后的无机载体浸渍于含烷基铝的有机溶剂中,反应时间为1~12h,优选1~4h,反应温度为室温至180℃,优选45~120℃,然后升温至60~210℃下干燥,优选80~
150℃,干燥时间为2~12h,优选2~4h,干燥后的产物可转移至惰性气体保护下保存待用。
[0097]
c)将上述产物浸渍于含钒源的溶液进行反应负载,浸渍时间为1~24h,优选4~12h,浸渍温度为20~180℃,优选45~120℃,然后在60~150℃之间干燥,优选80~130℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,然后将干燥后混合物置于惰性气体保护下保存备用。
[0098]
d)将上述产物置于含有机铬源的溶液中进行浸渍反应负载,浸渍反应时间为1~24h,优选4~8h,浸渍温度为20~120℃,优选45~80℃,然后在60~150℃之间干燥,优选80~100℃,干燥时间2~24h,优选6~16h,干燥过程中也可以采用真空,干燥结束后即得所述催化剂。
[0099]
e)将上述产物置于含有机金属助催化剂的溶液中进行活化反应,所述有机金属助催化剂与铬元素的摩尔比为0~1000,优选为0-100,更优选0-50;活化反应时间为0.5~20h,优选1~2h,活化温度为室温~100℃之间,优选室温~60℃之间,然后在60~120℃之间干燥,优选80~100℃,干燥时间2~8h,干燥过程中也可以采用真空,干燥结束后即得所述具有活性的催化剂。干燥后的催化剂在惰性气体气氛保护下保存待用。
[0100]
作为一个实例,制备本发明催化剂的具体操作包括:
[0101]
将sio2载体置于流化床内进行高温焙烧,焙烧过程在氮气氛围下进行,焙烧温度为600℃。焙烧达到指定时间后(如2~4h),停止加热,并使干燥和部分脱羟基的sio2自然冷却至室温,然后收集保存在有惰性气体(如氮气)保护的环境下。将上述焙烧后的载体与一定浓度的甲基铝氧烷甲苯溶液共混反应,甲基铝氧烷以铝计的加入量相对催化剂总重量为1wt%,反应在80℃下进行,并施以连续搅拌,反应约2h后,升温至130℃干燥4~6h,干燥过程通以氮气,以加速干燥。干燥好的改性载体置于惰性气体保护下保存待用。将上述产物浸渍在含一定浓度vocl3正己烷溶液中,钒的加入量相对于催化剂总重量符合本发明的要求(例如v的加入量为0.2~3wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,用正己烷在一定温度下(如室温~60℃)洗涤催化剂,再于一定温度下(如60~120℃)干燥4~6小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。将上述负载了一定钒催化剂前驱体的产物浸渍在含一定浓度双环戊二烯基铬的正己烷溶液中,铬的加入量相对于催化剂总重量符合本发明的要求(例如cr的加入量为0.2~2wt%);在一定温度下(如45-80℃)连续搅拌反应一定时间(例如4~8小时)后,再于一定温度下(如60~120℃)干燥4~6小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。将上述催化剂加入到三乙基铝的正己烷溶液中(三乙基铝与催化剂前驱体中铬的摩尔比为30∶1),然后在一定温度下(如60℃)连续搅拌反应一定时间(例如2小时)后,再于一定温度下(如85℃)干燥2~4小时,干燥过程在惰性气体,如氮气、氩气和氦气,环境下进行。干燥好的产物在惰性气体保护下转移保存。
[0102]
上述的负载型双中心催化剂能够用于催化烯烃聚合,尤其适用于催化乙烯的均聚或乙烯与α-烯烃的共聚。
[0103]
所述的烯烃一般包含乙烯单体,还可以包含共聚单体;所述共聚单体可以是具有3~20个碳原子的α-烯烃,例如丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯、1-辛烯、1-壬烯、1-癸烯、1-十二碳烯、1-甲基-1-戊烯、4-甲基-1-己烯等,这些可以单独使用或两种或更多种组
合使用。所述共聚单体优选为1-丁烯、1-己烯和1-辛烯中的至少一种;当共聚单体存在时,共聚单体的量一般为0~20vol%,优选0~10vol%,共聚单体的加入量是基于聚合时其相对于溶剂的体积浓度计算。
[0104]
当本发明负载型双中心催化剂用于催化乙烯的均聚或乙烯与α-烯烃的共聚时,所生产的聚乙烯产品具有如下特点:进行乙烯均聚时分子量分布较宽,部分呈双峰分布;进行乙烯与α-烯烃的共聚时,分子量分布较宽,部分呈双峰分布,且含有较低分子量乙烯均聚物与较高分子量乙烯与α-烯烃的共聚物。
[0105]
本发明负载型双中心催化剂乙烯在催化烯烃聚合中,可根据需要添加有机金属助催化剂以及分子量调节剂等;所述有机金属助催化剂可以为有机铝化合物,例如为三烷基铝alr3、二烷基烷氧基铝alr2or、二烷基卤化铝alr2x、铝氧烷、乙基倍半铝氯化物中的至少一种,其中r是烷基。更例如为三乙基铝、三异丁基铝、二乙基乙氧基铝、一氯二乙基铝和甲基铝氧烷等。所述有机金属铝化合物的用量满足:铝元素和总铬元素的摩尔比为0-1000,优选0-70,更优选0-50;所述的分子量调节可为氢气。
[0106]
本发明负载型双中心催化剂在催化烯烃聚合中,其对聚合的方法没有任何特别的限制,当用于催化乙烯的均聚或乙烯与α-烯烃的共聚时,聚合方法可以包括气相聚合工艺、淤浆聚合工艺、悬浮聚合工艺、本体聚合工艺、溶液聚合工艺等在常规实施方案和聚合条件下实施。
[0107]
优选的,当使用淤浆聚合工艺进行聚合时,包括向反应釜内加入乙烯,然后加入溶剂和助催化剂(有机铝化合物)并任选地加入氢气和共聚单体,最后加入本发明的负载型双中心催化剂开始聚合。其中所使用的溶剂一般为本领域所公知的用于烯烃聚合的任何溶剂,可为具有3~20个碳原子的烷烃,例如丙烷、正丁烷、异丁烷、正戊烷、异戊烷、新戊烷、正己烷、环己烷、正庚烷、正辛烷等;这些溶剂可以单独使用或可以两种或更多种组合使用。所使用的溶剂优选为异丁烷、异戊烷、正己烷、环己烷、正庚烷等。
[0108]
更优选的,当采用传统的淤浆聚合工艺实施聚合时,具体操作如下:先将聚合反应釜进行真空加热除杂,然后置换为高纯氮气,反复操作三次,再用少量乙烯单体置换一次,并最后将反应釜内充满乙烯至微正压(0.12mpa);向反应釜内加入脱水脱氧处理后的精制溶剂如正庚烷,一定量的烷基铝作为助催化剂,在氢调和共聚实验中还需分别加入一定量的氢气和共聚单体,待乙烯压力调至1mpa,最后加入本发明的催化剂开始聚合反应;反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录,在一定温度下(例如35℃-100℃)反应进行一定时间(例如1小时)后,加入盐酸/乙醇混合溶液终止反应;聚合物经洗涤,真空干燥后称重并分析。
[0109]
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
[0110]
实施例中所用试剂如无特殊说明均可从市场常规购得。
[0111]
实施例中的各种聚合物性质根据以下方法测量:
[0112]
高温凝胶色谱(ht-gpc)
[0113]
重均分子量和分子量分布用高温凝胶色谱测定:本实验采用pl-220型高温凝胶渗透色谱仪(polymer laboratories公司)来测定聚乙烯分子量及其分子量分布。实验中以1,2,4-三氯苯为溶剂,在160℃下测定。采用窄分布聚苯乙烯作为标样的普适校正法处理数据。
[0114]
13
c高温核磁共振谱(ht-13
cnmr)
[0115]
聚合物的短支链含量采用高温核磁碳谱测定:本实验采用brukeravance iii 500型核磁共振仪来测定聚乙烯短支链含量。实验中以氘代对二氯苯为溶剂,在110℃下测定,采用聚乙烯主链上的碳信号(位移在30.00ppm)为内标计算短支链含量。
[0116]
实施例1:
[0117]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(al的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0118]
实施例2:
[0119]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。将上述干燥产物加入到三异丁基铝的正己烷溶液中(a1与cr的摩尔比为10)进行活化反应,活化反应温度为45℃,活化时间为1h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂在氮气保护下转移至无水无氧条件下保存。
[0120]
实施例3:
[0121]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝
的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。
[0122]
实施例4:
[0123]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到含有三氯氧钒和双环戊二烯基铬的正己烷溶液中进行回流浸渍负载反应(cr与v的负载量相对于催化剂总重量分别为1wt%和2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。
[0124]
实施例5-1至5-4:
[0125]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到指定焙烧温度时(实施例5-1至5-4的指定焙烧温度分别为300℃,400℃、600℃和800℃)恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0126]
实施例6-1至6-4:
[0127]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝
的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(实施例6-1至6-4的v的负载量相对于催化剂总重量分别为0.5wt%、1wt%、3wt%和5wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0128]
实施例7-1至7-4:
[0129]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(实施例7-1至7-4中cr的负载量相对于催化剂总重量分别为0.5wt%、2wt%、3wt%和4wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0130]
实施例8-1至8-3:
[0131]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(实施例8-1至8-3中al的负载量相对于催化剂总重量分别为0.5wt%、1.5wt%和2wt%),在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0132]
实施例9-1至9-2:
[0133]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到钒卤化物(实施例9-1至9-2中钒卤化物分别为四氯化钒和三溴化钒)正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0134]
实施例10-1至10-2:
[0135]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(al的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于有机铬源(实施例10-1至10-2的有机铬源分别为二芴基铬和二茚基铬)的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0136]
实施例11-1至11-2:
[0137]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度烷基铝的正己烷溶液中(实施例11-1与11-2采用的烷基铝分别为三甲基铝和三乙基铝,al的负载量相对于催化剂总重量均为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为
6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0138]
实施例12-1至12-2:
[0139]
取10g多孔无机载体(实施例12-1至12-2的多孔无机载体分别为sio2/tio2(含3wt%ti,比表面积320m2/g,孔容1.5ml/g,孔径23nm)和sio2/zro2(含5wt%zr,比表面积500m2/g,孔容1.7ml/g,孔径21nm))于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(al的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0140]
实施例13-1至13-4:
[0141]
称取实施例1至4中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0142]
实施例14-1至14-4:
[0143]
称取实施例5-1至5-4中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0144]
实施例15-1至15-4:
[0145]
称取实施例6-1至6-4中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应
釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0146]
实施例16-1至16-4:
[0147]
称取实施例7-1至7-4中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为ai/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0148]
实施例17-1至17-3:
[0149]
称取实施例8-1至8-3中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0150]
实施例18-1至18-4:
[0151]
称取实施例9-1及9-2和实施例10-1及10-2中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0152]
实施例19-1至19-2:
[0153]
称取实施例11-1及11-2中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,
待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0154]
实施例20-1至20-2:
[0155]
称取实施例12-1及12-2中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0156]
实施例21-1至21-4:
[0157]
称取实施例1中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂。对于实施例21-1至21-4,分别加入用量为al/cr=10、al/cr=20、al/cr=40和al/cr=50的三异丁基铝(tiba)作助催化剂(cr为加入催化剂的总铬摩尔量),再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0158]
实施例22-1至22-3:
[0159]
称取实施例1中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂。对于实施例22-1至22-3,分别加入用量为al/cr=30(cr为加入催化剂的总铬摩尔量)的三乙基铝(tea)、一氯二乙基铝(deac)和甲基铝氧烷(mao)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0160]
实施例23-1至23-4:
[0161]
称取实施例1中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。对于实施例23-1至23-4,分别往反应釜中注入10ml、30ml、50ml和70ml的1-己烯共聚单体。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度
恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0162]
实施例24-1至24-4
[0163]
称取实施例1中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内压入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。对于实施例24-1至24-4,分别往反应釜中注入0.03mpa、0.05mpa、0.1mpa和0.2mpa的高纯氢。随后调节乙烯分压至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0164]
对比实施例1
[0165]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。
[0166]
对比实施例2
[0167]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0168]
对比实施例3
[0169]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。
[0170]
对比实施例4
[0171]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将经焙烧处理过的载体浸渍于含一定浓度三异丁基铝的正己烷溶液中(a1的负载量相对于催化剂总重量为1wt%)在45℃下反应2h,随后升温至85℃,并恒温4h,以脱除正己烷溶剂,干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0172]
对比实施例5
[0173]
取10g sio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)于流化床中进行焙烧脱水除羟基处理,升温速率为5℃/min,当达到500℃时恒温2h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。将上述产物加入到三氯氧钒正己烷溶液中进行回流浸渍负载反应(v的负载量相对于催化剂总重量为2wt%),回流浸渍时间为4h,回流浸渍温度为85℃,随后用正己烷对浸渍物进行3次洗涤,每次洗涤用正己烷为100ml。接着停止回流,并升温至120℃,并恒温4h,以脱除正己烷溶剂。干燥结束后将产物在氮气保护下转移至无水无氧条件下保存。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中(cr的负载量相对于催化剂总重量为1wt%),浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0174]
对比实施例6
[0175]
取10gsio2(比表面积300m2/g,孔容1.3ml/g,孔径20nm)浸渍在偏钒酸铵的水溶液中(v的负载量相对于催化剂总重量为2wt%),连续搅拌浸渍约4h,浸渍温度为60℃,随后升温至120℃,继续搅拌干燥8h。将干燥的混合物置于流化床中从室温开始进行升温焙烧,升温速率为1℃/min,当达到500℃时恒温4h,焙烧结束后开始自然冷却至室温,并转移至无水无氧条件下保存。上述过程中室温至150℃采用氮气气氛,150~500℃的升温阶段采用干空气气氛,500℃的恒温阶段,采用干空气气氛,自然冷却时由500℃降低至300℃时为干空气气氛,当低于300℃时切换至氮气气氛。将上述干燥产物浸渍于双环戊二烯基铬的正己烷溶液中,浸渍温度为45℃,浸渍时间为6h,随后进行升温干燥,干燥温度为80℃,干燥时间为4h,浸渍和干燥过程施加搅拌,干燥结束后将催化剂前驱体在氮气保护下转移至无水无氧条件下保存。
[0176]
对比实施例7-1至7-6
[0177]
称取对比实施例1-6中催化剂前驱体100mg,并混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液进行聚合实验。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为al/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终
止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0178]
对比实施例8-1至8-5
[0179]
称催化剂前驱体100mg,其中对比实施例8-1至8-5分别对应取对比实施例1-5所制备的催化剂,并将称取的催化剂混合于10ml经过精制的正庚烷溶液中形成催化剂前驱体悬浊液。将2l不锈钢高压聚合反应釜进行真空加热除杂,并用高纯氮气抽排三次,最后向反应釜内充微量精制乙烯至0.12mpa。往反应釜中注入10ml的1-己烯共聚单体。接着依次向反应釜内加入900ml精制的正庚烷溶剂,加入用量为a1/cr(cr为总铬摩尔量)=30的三异丁基铝(tiba)作助催化剂,再加入100ml脱水脱氧精制后的正庚烷溶剂。调节乙烯压力至1mpa,待釜内温度恒定在80℃后,利用高压氮气将催化剂前驱体悬浊液压入聚合釜中开始反应。反应过程中在线采集单体乙烯的瞬时消耗量并由电脑记录。1h后加入盐酸/乙醇混合溶液终止反应。过滤后将所得聚合物在真空干燥箱中60℃下干燥4h后称重并分析。
[0180]
上述实施例以及对比实施例所得催化剂用于乙烯聚合反应结果见表1。
[0181]
表1实施例以及对比实施例所得催化剂用于乙烯聚合反应结果
[0182][0183]
实施例13-1至13-4所用催化剂采用了不同的制备工艺,由表1所示,铬源与钒源的浸渍负载顺序以及是否采用金属有机助催化剂进行预还原活化均对催化剂的乙烯聚合活性有一定程度的影响。实施例13-1与13-2说明催化剂预还原活化将使催化剂催化活性明显提升。实施例13-1、13-3与13-4说明分步负载铬源与钒源的催化剂的催化活性优于共同负载铬源与钒源的催化剂。
[0184]
由实施例13-1与14-1至14-4可知,多孔无机载体的焙烧温度对催化剂的乙烯聚合活性也有较明显影响,焙烧温度由300℃提升到800℃时,催化剂的活性呈现先升高后降低的规律,其中在600℃左右达到最大值。这可能是由于催化剂载体表面的羟基密度对铬源与钒源具有双重影响所致,一方面过高密度的羟基对活性中心有毒化左右,而过低密度的羟基将不利于铬源与钒源的负载。
[0185]
由实施例13-1、15-1至15-4,以及16-1至16-4可知,催化剂前驱体的负载量有一个最优范围,即当铬源或钒源的加入量增加时,催化剂乙烯聚合活性呈逐步上升的趋势,同时,随着加入量的提高,其催化剂催化活性提高幅度越来越小。这可能是由于载体的比表面
积有限,铬源或钒源过高的加入量将难以实现铬源与钒源的有效负载。另一方面,铬源与钒源的相对加入量对聚乙烯产品的分子量及分子量分布具有显著影响,一般钒源的相对加入量越高,聚合物的分子量越高,分子量分布越窄。这是由钒活性中心更擅长于合成更高分子量聚合物产品的特性造成的。
[0186]
由实施例13-1、17-1至17-3可知,用于载体改性的烷基铝加入量也有一个最优范围,以三异丁基铝为改性组分时,其最佳范围按a1重量计为催化剂重量的1~1.5wt%左右。
[0187]
由实施例13-1与18-1至18-4可知,铬源和钒源的种类对催化剂的催化活性也有一定的影响。采用钒氯化物作为钒源要优于采用钒溴化物作为钒源;采用更大体积配体的二茂铬作为有机铬源,其催化活性略低,同时聚合物的分子量略高。另外,载体改性用烷基铝的种类对催化剂的催化活性也有一定的影响,其中还原性较弱的三异丁基铝更优于还原性更强的三甲基铝和三乙基铝。
[0188]
由实施例13-1与20-1至20-2可知,多孔无机载体的组成和比表面积等对催化剂乙烯聚合活性有较明显影响,采用一定含量的金属氧化物改性硅胶作为载体时,其催化活性更高,聚合物分子量更低。同时,更高的载体比表面积有利于提高催化剂的催化活性。
[0189]
由实施例13-1与21-1至21-4可知,助催化剂(活化剂)与催化剂前驱体的摩尔比对催化剂的乙烯聚合活性及聚合产物有较明显影响,且存在一个最优浓度。这主要是因为活化剂不足,金属中心得不到有效活化,导致催化活性较低,而活化剂过量将导致金属中心被过度还原为无活性组分,使催化活性降低,因此需要一个合理的活化剂浓度使催化活性达到最大值。
[0190]
由实施例13-1与22-1至22-3可知,助催化剂种类对催化剂乙烯聚合活性及聚合物分子量和分子量分布影响显著,其中三乙基铝的活化效果明显较差,而一氯二乙基铝活化效果较好,且聚合物分子量明显更高,甲基铝氧烷的活化效果介于中间,且聚合物分子量较低,分子量分布较窄。
[0191]
由实施例13-1与23-1至23-4可知,该催化剂体系具备正的共聚单体效应,即加入少量的共聚单体将使催化剂活性明显提升。同时引入的共聚单体促进了链转移反应的发生,使聚合物的分子量略有下降。通过高温核磁表征可知共聚单体被有效地插入到聚乙烯链中,形成丁基短支链,且随着共聚单体浓度的增大,聚合物中丁基短支链的相对含量逐步提高,但是提高的幅度越来越小,说明该催化体系具备一个最优的共聚单体浓度加入范围。
[0192]
由实施例13-1与24-1至24-4可知,提高氢气分压将明显降低聚合物分子量,同时催化剂的聚合活性也随之明显降低,说明该催化体系具备较优的氢调敏感性。
[0193]
由实施例13-1与对比实施例7-1至7-4可知,本发明所涉及的双金属中心催化剂的催化活性明显高于对应的两种单金属中心催化剂的活性之和,说明两种金属中心之间存在良好的催化协同效应。同时,双金属中心催化剂中钒活性中心合成的聚合物分子量明显更高,分子量分布更宽,而有机铬活性中心的聚合物分子量则较低,且分子量分布很窄。
[0194]
由实施例13-1与对比实施例7-5可知,对载体进行改性剂改性可明显提升双金属催化剂的乙烯聚合活性,同时载体改性可一定程度地降低聚合物的分子量。
[0195]
由实施例13-1与对比实施例7-6(专利cn103626899a所公开催化剂)可知,本发明所涉及经载体改性的铬、钒双金属催化剂更优于专利cn103626899a所公开铬、钒双金属催化剂。
[0196]
由实施例23-1与对比实施例8-5可知,对载体进行改性剂改性后所得催化剂用于烯烃共聚,其共聚单体插入效率明显更高。通过实施例8-1至8-2和8-3至8-4可知,对于单金属催化剂而言,对载体进行改性剂改性可明显提高单钒催化剂的共聚单体插入效率,但是对单铬催化剂却几乎无影响。这说明载体改性组分主要通过促进双金属中心催化剂中钒中心的共聚单体插入效率来改善其整体插入效率。
[0197]
当然,本发明还可有其它多种实施例,在不背离本发明精神及其实质的情况下,熟悉本领域的技术人员可根据本发明作出各种相应的改变和变形,但这些相应的改变和变形都应属于本发明权利要求的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。