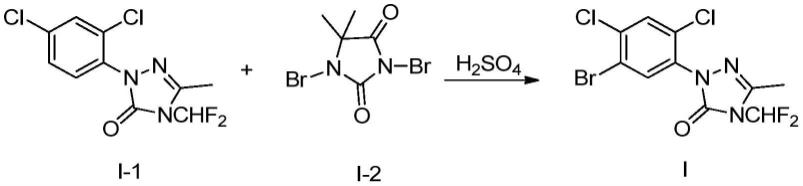
1.本发明涉及除草剂领域,具体涉及一种1,2,4-三唑-3-酮衍生物的2-(5-溴-2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮的制备方法。
背景技术:
2.卤代芳基二氟甲基三唑啉酮类化合物可作为医药或农药中间体化合物,基于卤代芳基二氟甲基三唑啉酮结构开发的除草剂如除草剂甲磺草胺已成功商业化,基于卤代芳基二氟甲基三唑啉酮的化合物的开发也得到了研究人员的重视。
3.2-(5-溴-2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮i是一种新的合成除草剂甲磺草胺的重要中间体化合物。
[0004][0005]
目前已报道的合成化合物i的方法主要包括如下几种方法。1)以碘为催化剂,溴化反应所用的溴化试剂为溴素、溴化氢或nbs反应制备得到化合物i。此方法涉及到溴化剂具有强腐蚀性和高毒性、双氧水具有强氧化性及易爆炸性、nbs原子利用率低等缺点,且产生大量“三废”,不利于工业化生产。2)以溴素为溴化剂,四氯化碳为溶剂制备化合物i,该工艺使用了溴素、四氯化碳等剧毒性试剂,危险性大,不利于该中间体的工业化放大生产。
技术实现要素:
[0006]
本发明的目的是为了克服现有技术存在的工艺危险、反应副反应多、转化不完全、副产难分离、产品纯度及收率偏低,“三废”量大等不利于工业化放大的问题,提供收率高,产品分离操作简便易行,易于工业化生产的方法。
[0007]
为了实现上述目的,本发明提供一种2-(5-溴-2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮的制备方法,其中,该方法包括以下步骤,
[0008]
1)在硫酸存在下,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲进行接触反应的步骤;
[0009]
2)将步骤1)得到的接触反应产物与水进行接触的步骤。
[0010]
优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲的摩尔比为1:0.4-1,优选为1:0.5-0.6,更优选为1:0.5-0.55。
[0011]
优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的摩尔比为1:0.5-20,优选为1:0.8-15,更优选为1:1-10。
[0012]
优选地,所述硫酸以硫酸水溶液形式使用;更优选地,所述硫酸水溶液的浓度为
10-98重量%,优选为20-98重量%。
[0013]
优选地,步骤1)中,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸混合后,控制温度在30℃以下,将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中,然后在该温度下反应1小时以上。
[0014]
优选地,在将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中之前,先将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的混合物进行冷却。
[0015]
优选地,所述冷却的温度为-15~25℃,优选为-10~20℃。
[0016]
优选地,步骤1)中,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸混合后,控制温度在-5~30℃,将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中进行反应,然后在该温度下反应1-5小时。
[0017]
优选地,步骤2)中,将所述接触反应产物缓慢添加到水中。
[0018]
优选地,所述水的用量是所述接触反应产物重量的0.5-5倍,优选为0.8-3倍,更优选为0.9-2倍。
[0019]
优选地,该方法还包括:将步骤2)的产物进行固液分离的步骤。
[0020]
根据本发明的方法,通过在硫酸存在下使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲进行接触反应,解决了现有工艺中存在强腐蚀、强氧化等危险工艺及副反应多、“三废”量大等问题,有利于工业化生产放大的实施。
具体实施方式
[0021]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0022]
本发明提供一种2-(5-溴-2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮的制备方法,其中,该方法包括以下步骤,
[0023]
1)在硫酸存在下,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲进行接触反应的步骤;
[0024]
2)将步骤1)得到的接触反应产物与水进行接触的步骤。
[0025]
根据本发明的方法,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮可以商购获得,也可以通过本领域通常的合成方法获得。
[0026]
根据本发明的方法,1,3-二溴-5,5-二甲基乙内酰脲的用量可以根据2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮的用量来选择,优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲的摩尔比为1:0.4-1;更优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲的摩尔比为1:0.5-0.6;进一步优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲的摩尔比为1:0.5-0.55。通过在上述范围内使用1,3-二
溴-5,5-二甲基乙内酰脲,可以进一步提高收率和得到的产物的纯度。
[0027]
根据本发明的方法,所述硫酸的用量可以根据2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮的用量来选择,优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的摩尔比为1:0.5-20,更优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的摩尔比为1:0.8-15,进一步优选地,2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的摩尔比为1:1-10。通过在上述范围内使用硫酸,可以进一步提高收率和得到的产物的纯度。
[0028]
在本发明中,优选所述硫酸以硫酸水溶液形式使用。在所述硫酸以硫酸水溶液形式使用时,其浓度可以在较大范围内变动,例如,所述硫酸水溶液的浓度可以为10-98重量%,优选为20-98重量%。
[0029]
根据本发明的方法,优选地,步骤1)中,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸混合后,控制温度在30℃以下,将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中,然后在该温度下反应1小时以上;更优选地,步骤1)中,使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸混合后,控制温度在-5~30℃,将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中进行反应,然后在该温度下反应1-5小时。
[0030]
在本发明中,在将1,3-二溴-5,5-二甲基乙内酰脲分批或持续加入到混合物中之前,优选先将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与硫酸的混合物进行冷却。作为所述冷却的温度例如可以为-15~25℃,优选为-10~20℃。
[0031]
根据本发明的方法,在所述接触反应完成后,仅需要将所述接触反应产物添加到水中后,通过搅拌降温即可析出目标产物,其后处理极其简单,非常适合工业制备。
[0032]
优选地,步骤2)中,将所述接触反应产物缓慢添加到水中,具体而言,可以控制添加时水体系的温度,例如可以在添加时控制水体系的温度为50℃以下,优选为40℃以下,更优选为30℃以下。
[0033]
根据本发明的方法,所述水的用量可以根据所述接触反应产物重量来选择,例如所述水的用量可以是所述接触反应产物重量的0.5-5倍;为了使目标产物更充分地析出,优选地,所述水的用量是所述接触反应产物重量的0.8-3倍;更优选地,所述水的用量是所述接触反应产物重量的0.9-2倍。
[0034]
根据本发明的方法,目标产物充分在水中充分析出后,通过固液分离来得到目标产物,所述固液分离没有特别的限定,可以为过滤或离心等本领域通常的固液分离方法。
[0035]
根据本发明的方法,在固液分离后,为了进一步提高目标产物的纯度,优选对固液分离得到的固相进行洗涤,所述洗涤的溶液例如可以为水。
[0036]
在洗涤后优选进行干燥,所述干燥的条件没有特别的限定,例如可以40-50℃下干燥4-6小时。
[0037]
根据本发明的方法,通过在硫酸存在下使2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮与1,3-二溴-5,5-二甲基乙内酰脲进行接触反应,解决了现有工艺中存在强腐蚀、强氧化等危险工艺及副反应多、“三废”量大等问题,有利于工业化生产放大的实施。
[0038]
以下将通过实施例对本发明进行详细描述,但本发明并不仅限于下述实施例。
[0039]
如无特殊说明,本发明中所涉及的操作和处理方法属于本领域常规方法。
[0040]
如无特殊说明,本发明中采用的仪器为本领域常规仪器。
[0041]
如无特殊说明,使用的各种原料均来自商购。
[0042]
实施例1
[0043][0044]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,100.0g,购自岳阳市宇恒化工有限公司)和98重量%浓硫酸(300.0g)加入四口瓶,搅拌至完全溶解后降温,冷却至0℃,控制温度在-5~5℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,53.8g),加完后保温搅拌2小时,将反应液缓慢倾入450.0g水中,搅拌分散,降温至室温,过滤,用200ml的水对固相进行水洗后,在45℃下干燥5小时得119.9g化合物i(其氢谱和质谱数据如下:1hnmr(400mhz,cdcl3),δ7.79(s,1h,arh),7.51(s,1h,arh),7.18(t,j=58.1hz,1h,chf2)2.28(s,3h,ch3)。esi-lcms,m/z 371.9004[m h]
),含量98.6重量%,收率93.2%。
[0045]
实施例2
[0046]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,50.0g)和20重量%硫酸(150g)加入四口瓶,搅拌至完全溶解后降温,冷却至0℃,控制温度在-5~5℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,26.9g),加完后保温搅拌2小时,将反应液缓慢倾入300g水中,搅拌分散,降温至室温,过滤,用100ml的水对固相进行水洗后,在45℃下干燥5小时得60.7g化合物i,含量98.9重量%,收率94.6%。
[0047]
实施例3
[0048]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,50.0g)和90重量%硫酸(150g)加入四口瓶,搅拌至完全溶解后降温,冷却至0℃,控制温度在-5~5℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,26.9g),加完后保温搅拌2小时,将反应液缓慢倾入225g水中,搅拌分散,降温至室温,过滤,用80ml的水对固相进行水洗后,在45℃下干燥5小时得60.1g化合物i,含量97.8重量%,收率92.7%。
[0049]
实施例4
[0050]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,50.0g)和80重量%硫酸(150g)加入四口瓶,搅拌至完全溶解后降温,冷却至0℃,控制温度在-5~5℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,26.9g),加完后保温搅拌2小时,将反应液缓慢倾入225g水中,搅拌分散,降温至室温,过滤,用90ml的水对固相进行水洗后,在45℃下干燥5小时得60.3g化合物i,含量97.1重量%,收率92.2%。
[0051]
实施例5
[0052]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,50.0g)和98重量%浓硫酸(150g)加入四口瓶,搅拌至完全溶解后降温,冷却至-10
℃,控制温度在-10~-5℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,26.9g),加完后保温搅拌2小时,将反应液缓慢倾入225g水中,搅拌分散,降温至室温,过滤,用100ml的水对固相进行水洗后,在45℃下干燥5小时得60.2g化合物i,含量98.7重量%,收率93.7%。
[0053]
实施例6
[0054]
将2-(2,4-二氯苯基)-4-(二氟甲基)-2,4-二氢-5-甲基-3h-1,2,4-三唑-3-酮(化合物i-1,50.0g)和98重量%浓硫酸(150g)加入四口瓶,搅拌至完全溶解后降温,冷却至20℃,控制温度在20~30℃分批加入1,3-二溴-5,5-二甲基乙内酰脲(化合物i-2,26.9g),加完后保温搅拌2小时,将反应液缓慢倾入225g水中,搅拌分散,降温至室温,过滤,用100ml的水对固相进行水洗后,在45℃下干燥5小时得60.1g化合物i,含量97.3重量%,收率92.4%。
[0055]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。