一组胰腺炎导致adm病变的肠道菌群标志物及其筛选方法与应用
技术领域
1.本发明属于肠道微生物技术领域,具体涉及一组胰腺炎导致adm病变的肠道菌群标志物及其筛选方法与应用。
背景技术:
2.胰腺炎肠道微生物紊乱诱导细菌移位引发败血症和并发感染,刺激巨噬细胞产生大量炎症细胞因子,给机体造成“二次打击”,最终导致胰腺和胰周组织坏死,加重死亡风险。人体的肠道内栖生着大量共生微生物,由至少1,000种细菌组成,称为肠道菌群,其总数是人体细胞总数的10倍,达到1014个。肠道菌群维持着胃肠道的稳态,广泛参与肠道及全身各器官生理机能的调节。近年来的研究发现,很多感染、代谢和免疫性疾病都与肠道菌群密切相关;肠道菌群失衡能够加重胃肠道疾病的继发感染和全身炎症反应,尤其是胰腺疾病。有研究表明益生菌能够纠正肠道微生物紊乱,是治疗胰腺炎的方法之一,具有高效低毒低成本的特征。然而,以往的研究对益生菌的作用不够明确,甚至由于缺乏对特定菌株和剂量的认识而导致临床效果出现相反的情况。因此,更好地探究肠道微生物的多样性组成,为胰腺炎制定适当的预防和治疗策略是十分迫切的。
3.已有一些研究描述了胰腺炎动物模型和患者不同肠道微生物的种类和特征。然而,这些研究有几个局限性:
①
它们通常重点研究胰腺炎的一个肠道部位的微生物群;
②
结肠粘膜部位的微生物群与粪便微生物群落有着明显的不同,然而,关于结肠微生物的研究却很少;
③
胰腺炎肠道微生物的微生物组成和功能分析尚未得到充分的研究与探讨。另外,用于研究胰腺炎进展过程中肠道微生物群失衡的大鼠模型比临床模型具有重要的优势,例如广泛的组织取样包括结肠和小肠内容物。积极探究胰腺炎肠道差异菌群及其与疾病的关系是研究胰腺炎发病机制的关键。16s rdna位于原核细胞核糖体小亚基上,指的是基因组中编码核糖体16s rrna分子对应的dna序列,也就是16s rrna的编码基因。该基因全长约1542bp,由9个可变区和10个保守区组成(可变区为v1到v9),其中保守区反映了生物物种间的亲缘关系,而可变区则表明物种间的差异,且变异程度与细菌的系统发育密切相关,被认为是最适于细菌系统发育和分类鉴定的指标。
4.目前,临床实践指南中建议使用外周血、尿淀粉酶和脂肪酶诊断胰腺炎并预测疾病严重程度。同时人们也在关注降钙素原(pct)、c-反应蛋白和hsa-mirna等实验室指标作为参数在预测胰腺炎adm病变中的作用。当前的诊断指标存在许多不足,包括准确性差、效率较低、耗时较长、有创性、操作复杂等,而且尚无有效的胰腺炎adm病变的肠道菌群预测标志物。
技术实现要素:
5.本发明是为了解决现有技术所存在的上述问题,基于16s rdna测序技术的微生物组学对胰腺炎大鼠模型的肠道不同部位,包括结肠、小肠和粪便的微生物群落进行分析,寻
dsdnaassay kit在promega quantifluor荧光定量系统上对文库进行定量,合格的文库浓度应在2nm以上,将合格的各上机测序文库梯度稀释后,根据所需测序量按相应比例混合,并经naoh变性为单链进行上机测序;使用miseq reagent kit v3(600cycles)试剂在illumina miseq测序仪进行2
×
300bp的双端测序。
18.(2.4)测序数据分析
19.①
数据拆分:对测序获得的双端数据根据barcode信息对样品进行数据拆分;
②
数据拼接和过滤:根据overlap关系进行数据拼接,同时对序列质量进行q20和q30等质控(quality control,qc)和过滤,获得最终的clean data;
③
操作分类单元(operational taxonomic units,otus)聚类:为了提高分析效率并删除测序错误的序列,我们将相似性大于97%的序列聚类为otu,每个otu对应于一条不同的16s rrna序列;
④
多样性分析:根据不同样品中otu的丰度,分析微生物的多样性,包括对样品中含有otu数目(丰富度)和群落结构的稳定性(均匀度)的计算和评估,以及不同样品间的物种组成结构差异(β多样性分析),从而研究不同样品物种组成异同;
⑤
物种注释和统计分析:根据每个otu代表序列与16s rdna数据库(rdp和nt-16s)比对结果,对otu进行物种分类统计,获得不同分类水平(界、门、纲、目、科、属、种)物种丰度表。对样品在不同分类水平的具体物种组成分析,检验组间存在显著性差异的物种,从而找到区分或影响不同样品(分组)的重要菌群;
⑥
微生物功能预测分析:我们采用stamp软件对微生物进行cog(clusters oforthologous groups)和kegg(kyoto encyclopedia of genes and genomes)分析,从而把物种的“身份”和它们的“功能”对应起来。
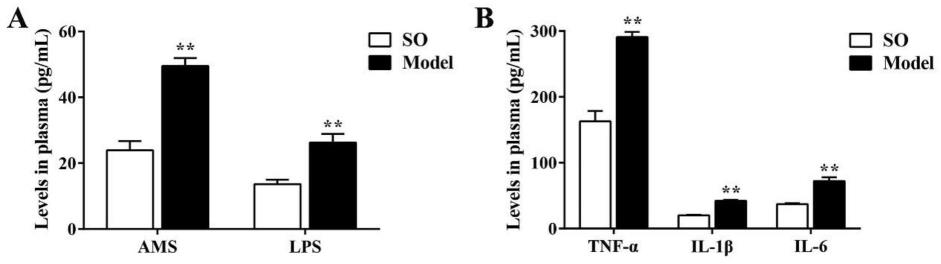
20.一组胰腺炎导致adm病变的肠道菌群标志物的应用,可以应用在制备胰腺炎愈后监测检测试剂盒中,或应用在筛选治疗或预防胰腺炎的药物中。
21.本发明与现有技术相比的有益效果是:
22.本发明提供了一种与胰腺炎到adm病变相关的微生物标志物,其中所述的微生物标志物包括:微生物标志物1乳酸杆菌lactobacillus在model大鼠小肠;微生物标志物2孪生球菌gemella和微生物标志物3土孢子杆菌terrisporobacter在model大鼠结肠、以及微生物标志物4拟杆菌属bacteroides、微生物标志物5大肠杆菌escherichia及微生物标志物6梭状芽孢杆菌clostridiumsensustricto,在model大鼠粪便样本中显著增加,其增加幅度均大于4.8倍,这些微生物标志物与促进胰腺炎到adm的出现起重要作用。
23.本发明的微生物标志物1lactobacillus(乳酸杆菌属);微生物标志物2gemella(孪生球菌属);微生物标志物4bacteroides(拟杆菌属)与急性胰腺炎具有显著的相关性,是判断胰腺炎是否发生adm转化最为灵敏和特异的、先于临床病理学特征的标志物,对临床医生进行胰腺炎患者管理具有一定的指导作用。
附图说明
24.图1是胰胆管逆行注射5%牛磺胆酸钠成功诱导大鼠胆源性胰腺炎模型;
25.图2是200
×
放大倍数下胰腺组织h&e染色图,结果表明,model大鼠胰腺组织出现水肿、炎症及腺泡细胞空泡化等病理损伤,如图中箭头所示;
26.图3是h&e染色结果显示胰腺炎诱导大鼠胰腺组织出现adm病变图;
27.图4是门水平的差异微生物统计学分析结果;
28.图5是lda分析显示在属水平的差异微生物。
具体实施方式
29.下面通过具体实施例详述本发明,但不限制本发明的保护范围。如无特殊说明,本发明所采用的实验方法均为常规方法,所用实验器材、材料、试剂等均可从商业途径获得。
30.实施例1胰腺炎adm病变大鼠模型建立
31.12只sprague-dawley(sd)大鼠,体重180-220g,自由饮用标准实验室食物和水(使用前经过高压灭菌),并在24
±
2℃、65
±
5%湿度和12h光/暗循环的spf环境中饲养。一周之后,sd大鼠随机分成两组(n=6):假手术(sham operation,so)组和胰腺炎模型(model)组。按照如下方法进行胆源性胰腺炎模型的构建:大鼠禁食12h,自由饮水,腹腔注射戊巴比妥钠(40mg/kg)进行麻醉。麻醉成功后取仰卧位,固定于鼠板,上腹部脱毛备皮,在无菌操作下行正中切口入腹,充分暴露术野,用小动脉夹夹闭肝门处胰胆管。于胰胆管汇合处逆行注射5.0%牛磺胆酸钠(0.1mg/100g体重)。作为对照,so组大鼠于胰胆管逆行注射等量无菌生理盐水。如图1所示,通过elisa法检测了ams和lps的含量,与so组相比,model组大鼠血浆中的ams和水平显著升高,炎症因子包括tnf-α、il-1β和il-6的分泌水平明显增加。此外,如图2所示,采用h&e染色观察胰腺炎胰腺组织的病理变化,so组大鼠胰腺无明显病理改变,而model组大鼠胰腺组织出现明显的损伤,具体表现为大面积间质水肿出血、中性粒细胞浸润和腺泡细胞空泡化等。如图3所示,h&e染色结果(400
×
放大倍数)观察到model组大鼠的胰腺组织中的部分腺泡细胞严重空泡化并形成导管状结构,即adm病变。因此,通过胰胆管逆行注射5%牛磺胆酸钠构建的胆源性胰腺炎adm模型是成功的,可用于后续的实验研究。
32.实施例2 16s rdna高通量测序
33.经过一系列的qc之后,从so和model大鼠的小肠和结肠内容物以及粪便等36个样本中一共获了得749,142个高质量的有效序列,所有样本的q30%平均值为94.98
±
2.30%。另外,长度小于200bp的序列数为45,917个,200~300bp为100,095个,300~400bp为246,016个,400~500bp为357,114个,分别占总数的6.13%、13.36%、32.84%和47.67%。因此,所有样本均符合建库要求,可用于后续分析。
34.为了探讨胰腺炎对肠道微生态的影响,我们比较了model大鼠和so大鼠不同肠道部位的微生物群落的组成。如图4所示,在门水平,胰腺炎大鼠小肠拟杆菌门(bacteroidetes)和结肠软壁菌门(tenericutes)的相对丰度均较so组显著降低(p《0.05)。此外,与so组相比,model组大鼠粪便样本中梭杆菌门(fusobacteria)和变形杆菌门(proteobacteria)的相对丰度显著增加,而软壁菌门(tenericutes)和蓝细菌门(cyanobacteria)的相对丰度明显减少(p《0.05)。此外,由于厚壁菌门(firmicutes)是所有样品中最丰富的门,我们比较了model和so大鼠粪便中厚壁菌门的丰度差异,结果表明,model组粪便中厚壁菌门数量较so组减少,但无统计学意义(p》0.05)。
35.为了寻找model组和so组间差异显著的微生物,我们进行了线性判别分析(linear discriminant analysis,lda),该分析是一种线性分类法,通常用于评估肠道微生物相对丰度的差异,可使用lefse软件分析获得,其中显著差异的logarithmic lda score设为2。我们计算了model大鼠与so大鼠菌群种属变化的lda评分,如图5所示,结果发现乳酸杆菌(lactobacillus)在model大鼠小肠、孪生球菌(gemella)和土孢子杆菌
(terrisporobacter)在model大鼠结肠、以及拟杆菌属(bacteroides)、大肠杆菌(escherichia)及梭状芽孢杆菌(clostridiumsensustricto)在model大鼠粪便样本中显著增加,这些差异菌群为胰腺炎的生物标志物。
36.以上所述实施方式仅为本发明的优选实施例,而并非本发明可行实施的全部实施例。对于本领域一般技术人员而言,在不背离本发明原理和精神的前提下对其所作出的任何显而易见的改动,都应当被认为包含在本发明的权利要求保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。