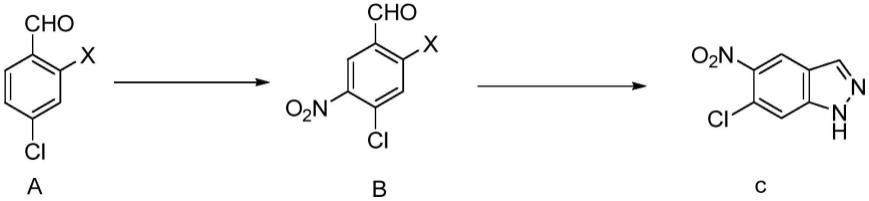
1.本发明属于药物合成领域,具体涉及一种制备6-氯-5-硝基-1氢-吲唑的方法。
背景技术:
2.冠状病毒(coronavirus)在系统分类上属冠状病毒科冠状病毒属,成熟的冠状病毒电镜下呈日冕状或者皇冠状,故被命名为冠状病毒,容易导致中枢神经系统疾病、普通感冒、下呼吸道感染和腹泻。严重急性呼吸系统综合征冠状病毒2型(sars-cov-2)是一种高致病性、大规模流行的人畜共患病毒。由sars-cov-2所引起的新型冠状病毒肺炎(covid-19)在全球大流行中,持续出现变异株,如德尔塔变异株(delta)和奥密克戎变异株(omicron),导致新冠疫苗保护能力减少甚至失效。
3.sars-cov-2病毒感染的症状从无症状疾病到中度和重度肺炎,以及危及生命的并发症,包括低氧性呼吸衰竭、急性呼吸窘迫综合征、多系统器官衰竭,并最终出现死亡。目前,防治病毒主要依赖于疫苗和药物。疫苗本身具有一定的局限性:免疫接种率低,很难产生有效的群体免疫;对高危人群很难产生有效的免疫力,如老年人、免疫缺陷人群;由于病毒rna聚合酶缺少翻译后校正机制,因此面对病毒持续突变时需不断进行新疫苗的研制,同时在快速流行早期难以在短时间内产生足够数量的新疫苗。因此,抗病毒药物研究仍然是抗病毒研究领域的热门话题。
4.日本盐野义制药公司报道的非共价mpro小分子抑制剂s-217662(cas:2647530-73-0)目前处于临床2/3期,该公司目前已经报道了部分积极临床研究结果。该化合物能够显著抑制包括α、β、γ和omicron等多种sars-cov-2变异株,表明其作为治疗新冠的治疗剂具有广泛的应用潜力。而且s-217622对一系列冠状病毒显示出广泛的抗病毒活性。大规模合成高收率、高纯度且低成本的s-217662具有重要意义。
[0005][0006]
6-氯-5-硝基-1氢-吲唑是一种合成s-217662的关键中间体。申请号为202210418619.9的中国专利申请公开了一种合成6-氯-5-硝基-1氢-吲唑的方法,路线如下所示。该方法以化合物1为起始原料,将化合物1混悬于水(80ml)和浓盐酸(25ml,0.3mol)中,0℃下,将nano2水溶液(6.9g,0.1mol)滴加至上述溶液中,然后搅拌20min,过滤,预冷的nabf4(12.g,0.1mol)的水溶液(40ml)加入到上述滤液中,搅拌40min。停止搅拌,过滤,冷乙醇和乙醚洗涤滤饼,收集滤饼、干燥制得重氮盐(11.5g,0.05mol)。将重氮盐溶于chcl3(100ml),将koac(8.15g,0.78mol)加入到上述溶液中,室温下搅拌2h,反应完全,停止搅拌。加水(50ml)淬灭反应,dcm萃取(50ml
×
3),合并有机相,饱和食盐水洗涤,无水na2so4干燥,过滤,浓缩,重结晶制得化合物2,即6-氯-5-硝基-1氢-吲唑。
[0007][0008]
但是,上述方法存在以下问题:(1)上述方法采用的起始原料(即化合物1)价格昂贵,高达5500元/公斤,增加了生产成本;(2)上述方法涉及的重氮化反应属于高危工艺,不适合工业化生产;(3)上述方法采用的nano2易产生难以除去的基因毒性杂质;(4)此外,本发明的发明人重复上述方法发现,其收率仅35%,远远达不到其声称的80%。
[0009]
为了克服上述问题,亟需开发出一种低成本、高收率、高纯度、适合工业化生产的方法来制备6-氯-5-硝基-1氢-吲唑。
技术实现要素:
[0010]
本发明的目的在于提供一种低成本、高收率、高纯度、适合工业化生产的制备6-氯-5-硝基-1氢-吲唑的方法。
[0011]
本发明提供了一种制备6-氯-5-硝基-1氢-吲唑的方法,所述方法包括以下步骤:
[0012][0013]
(1)化合物a与硝酸发生硝化反应,得到化合物b;x为卤素;
[0014]
(2)化合物b与水合肼反应,得到化合物c,即6-氯-5-硝基-1氢-吲唑。
[0015]
进一步地,x为氯或氟,优选为氟。
[0016]
进一步地,步骤(1)中,所述硝化反应是在催化剂的作用下进行的,所述催化剂为浓硫酸;化合物a、硝酸和催化剂的当量比为1:(3~4):(2~3);所述硝酸为浓硝酸;
[0017]
所述硝化反应的溶剂为浓硫酸,化合物a与溶剂的质量为1:(5~30);
[0018]
所述硝化反应的温度为-30~30℃,时间为1~60min。
[0019]
进一步地,化合物a、硝酸和催化剂的当量比为1:3.5:2.8;
[0020]
化合物a与溶剂的质量比为1:(12~20);
[0021]
所述硝化反应的温度为-20~25℃,时间为30min。
[0022]
进一步地,步骤(1)中,所述硝化反应结束后,还包括以下后处理步骤:将反应液倒入冰水中搅拌,加二氯甲烷萃取,取有机相用饱和碳酸氢钠溶液洗涤,干燥,得到化合物b。
[0023]
进一步地,步骤(2)中,化合物b和水合肼的质量比为(0.20~1.50):1;
[0024]
所述反应的溶剂为有机溶剂;
[0025]
所述反应的温度为50~70℃,时间为2~7h。
[0026]
进一步地,步骤(2)中,化合物b和水合肼的质量比为(0.44~1.03):1;
[0027]
所述有机溶剂为二甲基亚砜或n,n-二甲基甲酰胺;
[0028]
所述反应的温度为60℃,时间为4h。
[0029]
进一步地,步骤(2)中,所述水合肼为40%水合肼。
[0030]
进一步地,步骤(2)中,所述反应是在i2的存在下进行的,所述化合物b和i2的质量比为(4~6):0.01,优选为2.2:0.01。
[0031]
进一步地,步骤(2)中,所述反应结束后,还包括以下后处理步骤:在反应液中加入水,过滤,收集固体,干燥,得到化合物c。
[0032]
本发明中,“浓硫酸”指质量分数大于或等于70%的硫酸水溶液,实施例采用的市售浓硫酸密度为1.84g/ml;“浓硝酸”指浓度为8mol/l以上的发烟硝酸,实施例采用的市售浓硝酸的质量分数为98%。
[0033]
与现有技术相比,本发明制备6-氯-5-硝基-1氢-吲唑的方法具有以下有益效果:
[0034]
(1)本发明方法中采用的起始原料易得,可从市售商品中获得,价格便宜;
[0035]
(2)本发明方法操作简单,安全,且不涉及高危工艺,适合工业化生产;
[0036]
(3)本发明方法采用的原料安全无毒,没有使用nano2,不会产生难以除去的基因毒性杂质;
[0037]
(4)本发明方法制备得到的产物6-氯-5-硝基-1氢-吲唑纯度高,收率高,应用前景广阔。
[0038]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0039]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
[0040]
图1:实施例1所得6-氯-5-硝基-1氢-吲唑的1h nmr谱图。
具体实施方式
[0041]
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
[0042]
实施例1:制备6-氯-5-硝基-1氢-吲唑的方法
[0043][0044]
步骤一:将化合物a(7.5g,1eq)溶于浓硫酸(50ml,92g),搅拌,降温至-20℃。保持在-20℃下,滴加浓硫酸(11.77g,2.8eq)和浓硝酸(9.45g,3.5eq)的混酸,滴完后保持-20℃搅拌反应30min,然后在搅拌状态下恢复至室温,将反应液倒入冰水中搅拌,加dcm(二氯甲烷,3*20ml)萃取,取有机相用饱和碳酸氢钠溶液洗涤,再用无水硫酸镁干燥,过滤除去硫酸镁,将滤液旋蒸除去溶剂,得到中间产物化合物b(2,4-二氯-5-硝基-苯甲醛)。收率95%,纯
度89.9%。
[0045]
步骤二:向50ml三口瓶中加入步骤一制备的化合物b 2.2g,碘(i2)0.01g,dmso(二甲基亚砜)10ml搅拌溶解。再加入40%水合肼5g,搅拌均匀。加热并控制温度为60℃,用薄层色谱监测反应进展,4小时后反应完全。在反应液中加入水100ml,过滤,收集黄色固体,干燥,得到化合物c(即6-氯-5-硝基-1氢-吲唑)。收率56%,纯度98.6%。1h(nmr,d
6-dmso)=8.68(s,1h),8.37(s,1h),7.93(s,1h)。
[0046]
可以看出,利用实施例1的方法制备6-氯-5-硝基-1氢-吲唑的产物总收率为53.2%,纯度为98.6%。
[0047]
实施例2:制备6-氯-5-硝基-1氢-吲唑的方法
[0048][0049]
步骤一:将化合物a(7.5g,1eq)溶于浓硫酸(50ml,92g),搅拌,降温至0℃。保持在0℃下,滴加浓硫酸(11.77g,2.8eq)和浓硝酸(9.45g,3.5eq)的混酸,滴完后保持0℃搅拌反应30min,然后在搅拌装填下恢复至室温,将反应液倒入冰水中搅拌,加dcm(二氯甲烷,3*20ml)萃取,取有机相用饱和碳酸氢钠溶液洗涤,再用无水硫酸镁干燥,过滤除去硫酸镁,将滤液旋蒸除去溶剂,得到中间产物化合物b(2,4-二氯-5-硝基-苯甲醛)。收率96%,纯度78%。
[0050]
步骤二:向50ml三口瓶中加入步骤一制备的化合物b 2.2g,碘(i2)0.01g,dmso(二甲基亚砜)10ml搅拌溶解。再加入40%水合肼5g,搅拌均匀。加热并控制温度为60℃,用薄层色谱监测反应进展,4小时后反应完全。在反应液中加入水100ml,过滤,收集黄色固体,干燥,得到化合物c(即6-氯-5-硝基-1氢-吲唑)。收率42%,纯度94.1%。1h(nmr,d
6-dmso)=8.68(s,1h),8.37(s,1h),7.93(s,1h)。
[0051]
可以看出,利用实施例2的方法制备6-氯-5-硝基-1氢-吲唑的产物总收率为40.32%,纯度为94.1%。
[0052]
实施例3:制备6-氯-5-硝基-1氢-吲唑的方法
[0053][0054]
步骤一:将化合物a(17g,1eq)溶于浓硫酸(187ml,344.08g),搅拌,控温在25℃。保持在25℃下,滴加浓硫酸(29.8g,2.8eq)和浓硝酸(24.2g,3.5eq)的混酸,滴完后保持25℃搅拌反应30min,然后在搅拌装填下恢复至室温,将反应液倒入冰水中搅拌,加dcm(二氯甲烷,3*25ml)萃取,取有机相用饱和碳酸氢钠溶液洗涤,再用无水硫酸镁干燥,过滤除去硫酸镁,将滤液旋蒸除去溶剂,得到中间产物化合物b(2,4-二氯-5-硝基-苯甲醛)。收率99%,纯
度99%。
[0055]
步骤二:向50ml三口瓶中加入步骤一制备的化合物b 2.2g,碘(i2)0.01g,dmso(二甲基亚砜)10ml搅拌溶解。再加入40%水合肼5g,搅拌均匀。加热并控制温度为60℃,用薄层色谱监测反应进展,4小时后反应完全。在反应液中加入水100ml,过滤,收集黄色固体,干燥,得到化合物c(即6-氯-5-硝基-1氢-吲唑)。收率85%,纯度98.1%。1h(nmr,d
6-dmso)=8.68(s,1h),8.37(s,1h),7.93(s,1h)。
[0056]
可以看出,利用实施例3的方法制备6-氯-5-硝基-1氢-吲唑的产物总收率为84.15%,纯度为98.1%。
[0057]
实施例4:制备6-氯-5-硝基-1氢-吲唑的方法
[0058][0059]
步骤一:将化合物a(17g,1eq)溶于浓硫酸(187ml,344.08g),搅拌,控温在25℃。保持在25℃下,滴加浓硫酸(29.8g,2.8eq)和浓硝酸(24.2g,3.5eq)的混酸,滴完后保持25℃搅拌反应30min,然后在搅拌装填下恢复至室温,将反应液倒入冰水中搅拌,加dcm(二氯甲烷,3*25ml)萃取,取有机相用饱和碳酸氢钠溶液洗涤,再用无水硫酸镁干燥,过滤除去硫酸镁,将滤液旋蒸除去溶剂,得到中间产物化合物b(2,4-二氯-5-硝基-苯甲醛)。收率》99%,纯度99%。
[0060]
步骤二:向50ml三口瓶中加入dmf(n,n-二甲基甲酰胺)9ml和40%水合肼(1.95g,3.2eq),然后降温至0℃,搅拌状态下加入步骤一制备的化合物b(2.0g,1.0eq)。加入完毕后,缓慢升温至室温持续搅拌2h,再升温至60℃,继续反应1h。用薄层色谱监测反应进展,4小时后反应完全。在反应液中加入水100ml,过滤,收集黄色固体,干燥,得到化合物c(即6-氯-5-硝基-1氢-吲唑)。收率90%,纯度95.9%。1h(nmr,d
6-dmso)=8.68(s,1h),8.37(s,1h),7.93(s,1h)。
[0061]
可以看出,利用实施例4的方法制备6-氯-5-硝基-1氢-吲唑的产物总收率为89.1%,纯度为95.9%。
[0062]
综上,本发明提供了一种制备6-氯-5-硝基-1氢-吲唑的方法。本发明方法中采用的起始原料易得,价格便宜;本发明的方法操作简单,安全,不涉及高危工艺,得到的产物6-氯-5-硝基-1氢-吲唑纯度高,收率高,适合工业化生产。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。