一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物及其制备方法
技术领域
1.本发明涉及配位化合物技术领域,具体涉及一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物及其制备方法。
背景技术:
2.近几十年来,氮氧自由基与过渡金属的磁性配合物因其丰富的结构和在功能材料中的潜在应用而备受关注。这些材料在自旋电子学、高密度信息存储、磁共振成像和双稳态智能开关器件方面显示出良好的应用前景。自意大利化学家dante gatteschi首次发现single-chain magnets(scm)以来,氮氧自由基更受到了广泛的关注。此后,人们对含氮氧自由基的磁性配合物进行了大量的研究。由于能耗小,易于合成,芳香族氮氧化合物(nitr)作为分子单元已被广泛应用于分子磁性材料的设计与合成。通过金属离子将吡啶、噻吩、咪唑等多种改性官能团引入ullman型氮自由基化合物中,能成为稳定的自旋载体。氮氧自由基配体上的各种取代不仅会导致配位模式的变化,而且会导致电子行为的变化。到目前为止,一些具有封闭结构的自由基配合物相继被研究。从磁性的角度来看,嘧啶-氧键和羧基桥连co(ii)配合物的磁性也得到了很好的认可,高自旋co(ii)离子含有3个未配对电子,具有较大的磁矩与氮氧化物自由基的组装倾向于形成单分子磁体。双核co(ii)配合物通常作为模型来理解结构参数对相邻磁性单元之间交换耦合作用的大小方面的影响。它们表现出有趣的磁弛豫行为。然而,由于很难对最终产品的性能和结构进行微调,氮氧自由基在结构设计及其功能的实现仍然是一个重大的挑战。天津工业大学周旭光公开了一种co(ii)-2-(5-溴-3-吡啶)-4,4,5,5-四甲基咪唑啉-3-氧化-1-氧基自由基配合物及其制备方法,通过溶液加热回流一小时等,而分子内电子的分布,磁作用效果没有提及。基于此,申请人设计、改变芳香换上吸电子取代基,为供电子的甲氧基,通过简便方法获得了基于甲氧基嘧啶nit-mo-pmy新型co(ii)磁性配合物,可以通过共轭体系中电子离域,导致中心原子与自由基配体的磁耦合作业,进而提升其磁性能。
技术实现要素:
3.本发明的目的是为解决上述技术问题及不足,提供一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物及其制备方法。
4.本发明解决上述技术问题,所采用的技术方案是:一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物,化学式为[co2(hfac)4(nit-mo-pmy)2],其中hfac为1,1,1,5,5,5-六氟乙酰丙酮,nit-mo-pmy为2-(2-甲氧基嘧啶基)-4,4,5,5-四甲基咪唑啉-3-氧化物-1-氧基自由基配体,该配合物晶体属三斜晶系,空间群为p-1,晶胞参数为:α=68.688(3)
°
,β=85.726(3)
°
,γ=72.990(3)
°
,晶胞体积z=2。
[0005]
在配合物的结构单元中,不对称单元包含一个co(ii)阳离子,两个nit-mo-pmy配体和两个hfac片段,co(ii)离子采用六配位的准八面体结构,周围提供了n1o5给体单元,在给体单元中,5个氧原子与co(ii)离子配位,其中4个氧原子来自hfac配体,1个来自氮氧自由基配体,另一个n原子来自氮氧自由基配体的嘧啶环。
[0006]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物的制备方法,包括以下步骤:
[0007]
1)、取自由基有机配体nit-mo-pmy,加入水与有机溶剂的混合溶液中,搅拌混合,得到溶液a;
[0008]
2)、取六氟乙酰丙酮钴盐加入水与有机溶剂的混合溶液中,搅拌混合,得到溶液b;
[0009]
3)、在溶液a中加入有机溶剂中搅拌均匀后得到溶液c,在静置条件下,逐滴滴加溶液b,再滴加有机溶剂定容,最后密闭反应容器并在黑暗处静置;
[0010]
4)、静置完成后,收集反应容器壁上析出的晶体颗粒,经洗涤、过滤及干燥,即得到磁性co的配合物。
[0011]
其中,有机溶剂为甲醇、乙醇、二氯甲烷或dmf,水与有机溶剂的混合溶液中水与有机溶剂的体积比为1:1~10。
[0012]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物的制备方法,包括以下步骤:
[0013]
1)、取四水合硝酸钴悬浮于六氟乙酰丙酮的无水乙醇溶液中,搅拌加热至120℃,经回流反应,获得晶态co(hfac)2·
4h2o;
[0014]
2)、将co(hfac)2·
4h2o悬浮于无水正庚烷中,然后将悬浮液加热至90℃进行反应;
[0015]
3)、当悬浮液变成透明的亮黄色溶液时,将温度降低至70℃,加入含自由基有机配体nit-mo-pmy的氯仿溶液,继续反应;
[0016]
4)、反应结束后,过滤得到深红色滤液,将其置于低温下缓慢挥发,得到紫红色晶体,将晶体洗涤、干燥后,即得到磁性co配合物。
[0017]
其中,步骤1)中回流反应具体为:搅拌加热到120℃,回流4小时。
[0018]
其中,步骤4)中将深红色滤液置于4℃温度环境下,缓慢挥发8天,得到紫红色晶体。
[0019]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物的制备方法,包括以下步骤:
[0020]
1)、取自由基有机配体nit-mo-pmy,加入水与有机溶剂的混合溶液中,搅拌混合,得到溶液a;
[0021]
2)、取四水合醋酸钴和六氟乙酰丙酮加入溶液a中,再将上述溶液加入水与有机溶剂的混合液中,搅拌均匀后得到溶液b;
[0022]
3)、将溶液b加入无水正庚烷中,用磁力搅拌器在常温常压下搅拌,然后加入稀naoh溶液调节ph值为7-8,得到前驱液c,备用;
[0023]
4)、将前驱液c加热至90℃进行反应,当悬浮液变成透明的亮黄色溶液时,将温度降低至70℃,加入含自由基有机配体nit-mo-pmy的氯仿溶液,继续反应;
[0024]
5)、反应结束后,过滤得到深红色滤液,将其置于低温下缓慢挥发,得到紫红色晶体,将晶体洗涤、干燥后,即得到磁性co配合物。
[0025]
其中,步骤5)中将深红色滤液置于4℃温度环境下,缓慢挥发10天,得到紫红色晶体。
[0026]
其中,有机溶剂为甲醇、乙醇或dmf,水与有机溶剂的混合溶液中水与有机溶剂的体积比为1:1~3。
[0027]
本发明具有以下有益效果:
[0028]
本发明的co配合物磁性材料中含有磁性金属离子和含有单自旋电子的自由基,结晶性较好,无毒、无污染,热稳定性高,分子内具有较强的磁耦合作用,制备便捷;制备设备简单,综合性能优良,适合磁性材料规模化生产,该材料可满足无人机工业,磁存储、智能材料,安全控制等方面。
附图说明
[0029]
图1是实施例1所制备产物样品实物照片;图2是实施例1所制备产物的红外光谱图;图3是实施例1所制备产物的粉末x-射线衍射(pxrd)图谱与单晶衍射数据模拟的xrd的比较图;图4是实施例1所制备产物的配合物分子的基本单元结构图;图5是实施例1所制备产物的配合物的晶体结构图;图6是实施例1所制备产物的摩尔磁化率的倒数随温度变化的曲线图;图7是实施例1所制备产物的摩尔磁化率与温度乘积与温度关系的曲线图。
具体实施方式
[0030]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。实施例1
[0031]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物及其制备方法,包括以下步骤:
[0032]
(一)、制备自由基有机配体nit-mo-pmy
[0033]
自由基有机配体2-(2-甲氧基嘧啶基)-4,4,5,5-四甲基咪唑啉-3-氧化物-1-氧基自由基制备委托实验室的有机合成专业老师,按照文献程序制备去完成,不在本专利保护之列。参考liux,feng x,meihaus kr,meng x,zhangy,li l,li ujl,pedersenks,kellerl,shiw,zhangyq,cheng p,long j r.angew.chem.int.ed.,2020,59(26):10610-10618等。
[0034]
自由基有机配体nit-mo-pmy的结构式如下:
[0035][0036]
(二)、制备co磁性材料
[0037]
a、取0.1mmol自由基有机配体溶液10ml水与甲醇的混合液中(水与甲醇的体积比为1:1),得到溶液a;
[0038]
b、取0.1mmol的六氟乙酰丙酮钴盐加入5ml的水与乙醇的混合液(水与乙醇的体积比为1:3),得到溶液b;
[0039]
c、将溶液a至于25ml的比色管中,加入20~30mldmf中搅拌均匀后得到溶液c,在静置条件下,用胶头滴管逐滴滴加溶液b,再滴加乙醚至20ml,加塞,用聚四氟乙烯胶带封闭管口在黑暗处静置1周。
[0040]
d、观察到晶体颗粒在比色管的管壁上析出(如图1所示)。
[0041]
e、按照上述方法洗涤、过滤、干燥。
[0042]
pxrd测试
[0043]
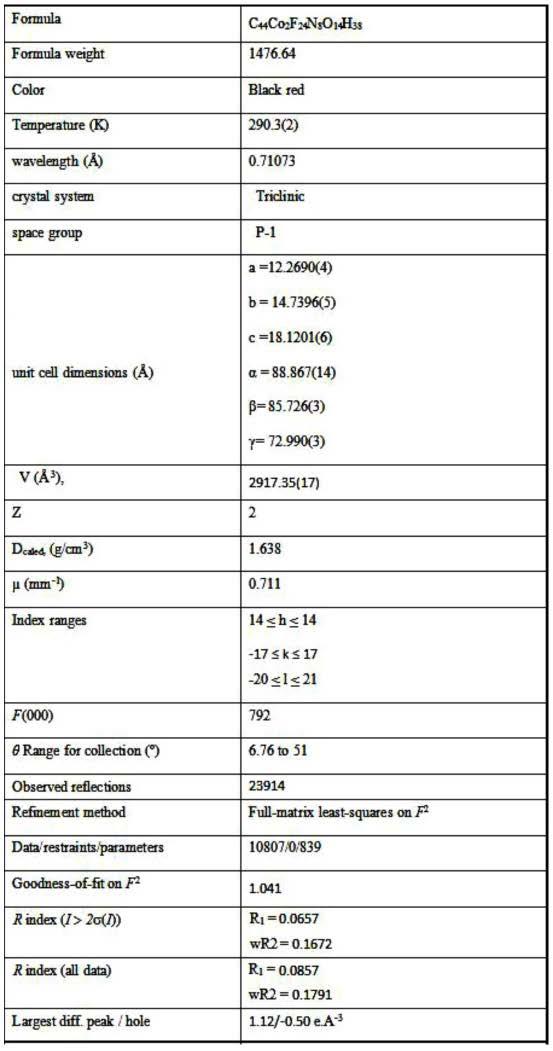
将所得产物[co2(hfac)4(nit-mo-pmy)2],用德国的bruker公司的smartapexii型单晶x射线衍射仪进行分析,如图3所示:发现合成最终产物的pxrd图谱与单晶衍射数据模拟的xrd图谱几乎全部吻合,在2θ分别为6.7,9.8,12.6,14.9,23.6,25.2
°
等处出现强的特征衍射峰,同时得到产物单晶,标题配合物晶体结构测试的实验条件、数据收集、结构解析和修正方法以及晶体学数据在表1中列出。
[0044]
表1:产物化合物晶胞数据及晶体精修细节
[0045][0046]
配合物的化学式未c
44
co2f
24
n8o
14h38
,通过diamond 3d晶体画图软件绘出产物的晶体结构,如图4所示。分析co(ii)配合物x-射线单晶衍射的数据后发现,该标题配位化合物属于三斜晶系,处在p-1空间群,其晶胞参数为α=68.688(3)
°
,β=85.726(3)
°
,γ=72.990(3)
°
,v=2917.35(17)a3,z=2。如图1所示,在配合物的结构单元中,不对称单元包含一个co(ii)阳离子,两个nit-mo-pmy配体和两个hfac片段。co(ii)离子采用六配位的准八面体结构,周围提供了n1o5给体单元。在给体单元中,5个氧原子与co(ii)离子配位,其中4个氧原子来自hfac配体,1个来自氮氧自由基配体,另一个n原子来自氮氧自由基配体的嘧啶环。co(ii)离子仅跟嘧啶环的一个n原子配位。与周旭光公开的结构不一致。hfac的三个氧原子和氮氧自由基的一个n原子位于赤道面,嘧啶部分的n原子占据了轴位,从而完成了
八面体配位的几何结构。其中氮氧自由基通过嘧啶环上的一个氮原子和氮氧自由基基团上的一个氧原子形成双齿配体,并以双齿配体的形式与co(ii)结合,形成八面体配位构型的二聚体配合物,成为闭环结构的氮氧自由基类配位化合物,如图5所示,co-o距离范围从2.114(3)到o-co-o角从78.79(13)到171.09(12)
°
不等。此外,n-o距离在1.227和之间。。在co(ⅱ)配合物中,由原子c(1)、c(3)、c(4)、c(5)和n(4)、n(3)组成的嘧啶平面与相邻的嘧啶平面晶体学上完全相同,并相互平行,平面间距离为这样的面间距离表明co(ⅱ)配合物中两个芳香环之间没有明显的π-π堆积。分子co(ii)
···
co(ii)距离为小于分子间最近的co(ii)
···
co(ii)距离(约)。最近的n-o
··
o-n距离为值得注意的是,co(1)-o(5)中参与自由基配体的键为比co(1)-o(1)的短,表明自由基配体与中心离子的相互作用更强。
[0047]
在2.0-300k的范围内,在1koe磁场作用下,研究配合物的磁性能,测试磁化率的变化。研究发现40-300k测试温度的范围内,其摩尔磁化率的倒数符合居里-韦斯定律,如图6所示。呈现负的韦斯温度为-36.27k,居里常数c=4.46cm3kmol-1
,初步表明配合物中存在整体化化合物内存在反铁磁相互作用。
[0048]
图7是本发明实施例1所制备产物[co2(hfac)4(nit-mo-pmy)2]的摩尔磁化率与温度乘积与温度关系的曲线图。
[0049]
如图7所示,在300k下,χmt的乘积为3.32cm3kmol-1
,略高于一个高自旋co(ii)离子(s=3/2)和一个有机自由基(s=1/2)在没有交换作用的情况下co(ii)离子与一个有机自由基(s=1/2)的期望自旋值(2.25cm3mol-1
k)。随着温度的下降,χmt值在最低2k时平稳地下降到0.46cm3kmol-1
。这表明co(ii)离子与氮氧配体之间存在反铁磁作用耦合。通过磁性能进行拟合,结果为j=
–
7.280325cm-1
,g=2.088,r=7.712
×
10-5
。如图7表明,这种化合物存在较强的分子内反铁磁偶联。实施例2
[0050]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co配合物及其制备方法,包括以下步骤:
[0051]
(一)、制备自由基有机配体nit-mo-pmy(方法同实施例1)。
[0052]
(二)、制备前驱液
[0053]
a、co(no3)2·
4h2o(24.9mg,0.1mmol)悬浮于25ml六氟乙酰丙酮的无水乙醇溶液中,搅拌加热到120℃,回流4小时,趁热过滤,冷却,获得晶态co(hfac)2·
4h2o。
[0054]
b、将co(hfac)2·
4h2o(48.9mg,0.1mmol)悬浮于25ml无水正庚烷中,然后将悬浮液加热至90℃,在此温度下反应1h。
[0055]
c、当悬浮液变成透明的亮黄色溶液时,将温度降低至70℃左右,加入10ml含nit-mo-pmy(26.6mg,0.1mmol)的氯仿溶液。
[0056]
d、晶态化合物的制备:0.5h后,过滤得到深红色滤液,将其放入4℃的冰箱中,缓慢挥发8天,得到紫红色晶体。粗产品首先使用蒸馏水洗涤,然后置于真空干燥器中干燥24h。产率:37mg,收率为48%(基于cobalt元素计算)。对材料c、h、n、元素组成含量进行分析:化学式为c
44
co2f
24
n8o
14h38
,理论计算元素含量百分比(calcd.):c35.79,h2.59,n7.59:实际测定(found):c35.68,h3.53,n7.53。
[0057]
(三)、制备磁性材料
[0058]
e、对得到的粉体真空干燥,研磨均匀,称重。实施例3
[0059]
一种基于甲氧基嘧啶氮氧自由基配体的磁性co(ii)配合物及其制备方法,包括以下步骤:
[0060]
(一)、制备自由基有机配体nit-mo-pmy(方法同实施例1)。
[0061]
(二)、制备磁性材料
[0062]
a、取0.1mmol的实施例1步骤(一)的自由基有机配体溶于40ml水与有机溶剂的混合液中,得到溶液a;
[0063]
b、取0.1mmol的四水合醋酸钴(e,co(ch3coo)2·
4h2o)和0.1mmol的六氟乙酰丙酮(cf3coch2cocf3)加入10ml的溶液a中,加入20~50ml水与甲醇的混合液中(水与甲醇的体积比为1:2),搅拌均匀后得到溶液b;
[0064]
c、将溶液b加入无水正庚烷中,用磁力搅拌器在常温常压下搅拌,然后加入稀naoh溶液调节ph值为7-8,得到前驱液c,备用。
[0065]
d、然后将悬浮液加热至90℃,在此温度下反应2h。当悬浮液变成透明的亮黄色溶液时,将温度降低至70℃左右,加入10ml含nit-mo-pmy(26.6mg,0.1mmol)的氯仿溶液。0.5h后,过滤得到紫红色滤液,将其放入4℃的冰箱中,缓慢挥发10天,得到适合x-射线单晶衍射的晶体。这些产品首先使用蒸馏水洗涤,然后置于真空干燥器中干燥24h。产率:36mg(49%)。
[0066]
e、将步骤d中制得的粉体干燥研成粉末。
[0067]
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变形或修改,这并不影响本发明的实质内容。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。