1.本发明涉及氢化工业催化剂制备技术领域,具体涉及一种用于强力霉素生产的新型钯碳催化剂的制备方法。
背景技术:
2.目前强力霉素的生产工艺大多是以土霉素为原料,经氯代、脱水、氢化、转化精制而得。强力霉素生产工艺中,中间体11α-氯-6-次甲基土霉素经过手性不对称催化氢化合成强力霉素是整条工艺路线中最为重要的一步,也是对产品质量与产量影响最大的一环。现在工业上主流的氢化路线是使用“一锅法”,中间体11α-氯-6-次甲基土霉素在催化剂的催化作用下一步反应生成α-6-多西环素即目标产物强力霉素,但由于6号位碳原子是手性碳原子,同时还会有β构型的副产物产生,因此催化剂的活性和选择性至关重要。
3.目前,工业上最主流的催化剂一种是毒化钯碳,另一种是三苯基膦氯化铑。其中,三苯基膦氯化铑是一种均相催化剂,带来了高选择性和高反应活性的同时也带来了无法回收套用的难题,同时又因为金属铑价格高昂导致成本更高。《浅谈盐酸多西环素的氢化合成》(中文科技期刊数据库(全文版).工程技术.2016年第11月.麻伟创等)提出在盐酸多西环素的氢化合成生产中采用可回收的以硅胶固定的均相铑催化剂,提供了一种提高铑套用次数的思路,但并未解决铑价格高昂导致的高成本问题。中国专利cn101352692a公开了一种生产药品多西环素的氢化工艺铱催化剂及其应用,主要介绍了一种铱催化剂,其可以达到与铑催化剂相当的活性及选择性,然而同样未解决套用问题,不适用于工业化。
4.毒化钯碳相较于铑催化剂虽然选择性较差但是可以多次重复使用,且钯价格远低于铑,在工业上使用更普遍。中国专利cn110918121a公开了一种强力霉素生产用氢化催化剂及其制备方法和应用,提出了聚苯乙烯为载体的钯基催化剂,应用于合成强力霉素,但相较于毒化钯碳其成本更高,且收率和选择性并不足够优越到替换毒化钯碳。中国专利cn102086165a公开了一种pd催化剂在生产强力霉素氢化工艺中的应用,通过选用聚氯乙烯多乙烯多胺负载钯配合物(pvc-pp-pd)为催化剂,溶于有机溶剂中进行催化反应,在不使用毒剂的情况下便达到了较高的选择性,但对比毒化钯碳工艺成本还是高,且涉及到高分子聚合反应,存在较大的安全风险。中国专利cn107056641a公开了一种强力霉素的制备方法,使用无机负载化合物碳酸钙或羟基磷灰石制备的钯基催化剂用于强力霉素生产工艺的氢化反应,提高了收率,降低了工业生产成本,然而载体的耐腐蚀性差,对于11α-氯-6-次甲基土霉素对甲苯磺酸盐加氢还原反应而言的酸性ph,其很容易发生载体的溶解并造成贵金属的流失。
5.工业上常用的毒化钯碳的载体活性炭通常是直接从活性炭生产厂家直接购买后使用,这些活性炭基本上都是用椰壳、竹子、果核等木质材料经炭化、活化(氯化锌法、氢氧化钾法等)得到内外孔道一致的活性炭。该活性炭确实适合用作催化剂载体,但对于11α-氯-6-次甲基土霉素催化氢化反应来讲,基本上只是活性炭壳层的钯在起催化作用,深处孔道的钯得不到有效利用,且催化剂生产过程中常用的抑制剂甲基硫氧嘧啶对环境污染毒化
作用很大,亟需替代。
技术实现要素:
6.本发明所要解决的技术问题是:针对现有技术存在的不足,提供一种用于强力霉素生产的新型钯碳催化剂的制备方法,催化剂的催化效果好,生产成本低,对环境更加友好。
7.为解决上述技术问题,本发明的技术方案是:一种用于强力霉素生产的新型钯碳催化剂的制备方法,包括以下步骤:a:木质材料用纯化水清洗后用乙醇浸泡2h,120℃烘干至恒重后粉碎成颗粒,然后在600℃马弗炉中氮气氛围下炭化1.5h得炭化固体;b:炭化固体用浓度为1 mol/l的盐酸溶液预浸渍7~9h,过滤,并用清水冲洗表面后转入质量分数为20%的氢氧化钾溶液中浸渍1h,再次过滤,固体于70℃烘箱中干燥至恒重,并在750~850℃马弗炉中氮气氛围下活化2h;c:活化后的固体用80~85℃热水打浆4~6h,过滤淋洗后得活性炭载体;d:将活性炭载体放入反应釜中,加入纯化水,反应釜的加热温度为30℃,反应釜的搅拌转速为100 rad/min,得到的活性炭浆液持续打浆备用;e:向钯质量含量为0.5%的氯钯酸溶液中滴入质量分数为15%的羧酸盐溶液,其中羧酸盐中羧酸根和钯元素的摩尔比为10:1,然后滴加碱液调节ph到7.25~8,得混合溶液;f:将混合溶液滴入步骤d中的反应釜中,控制反应釜的加热温度为30℃,搅拌转速为100 rad/min,滴加完成后继续搅拌12h,然后调节搅拌转速为300rad/min,并向反应釜内加入质量分数为1%的苯并噻唑乙醇溶液,其中苯并噻唑与氯钯酸溶液中钯元素的摩尔比为1:5,反应釜进行氮气置换,再用0.50mpa氢气置换三次,在1.2mpa氢气压力下向反应釜内加氢反应5~7h,抽滤后滤饼用水打浆4~6次,每次0.5h,过滤后得钯碳催化剂。
8.优选的,步骤b中炭化固体与盐酸溶液的质量比为1:5~25。
9.优选的,炭化固体与氢氧化钾溶液的质量比为1:5~25。
10.优选的,步骤e中的碱液为碳酸氢钠或碳酸钠中的一种。
11.优选的,步骤e中碱液的滴加时间为0.9~1.2h。
12.优选的,步骤e中羧酸盐溶液的滴加时间为1~5min。
13.优选的,步骤f中混合溶液的滴加时间为1.5~2 .5h。
14.优选的,步骤f中氮气置换为在0.50 mpa氮气置换空气三次。
15.优选的,步骤a中的木质材料为晾晒干燥后的果核或椰壳、竹子等。
16.由于采用了上述技术方案,本发明的有益效果是:1、通过乙醇浸泡、炭化、活化、热水打浆及过滤淋洗得到的炭载体壳层孔道发达,且内核孔道小而少,然后通过浸渍法负载钯得到了蛋壳型的钯碳催化剂,钯的有效利用率更高。
17.2、因为活性炭载体壳层孔道发达,且内核孔道小而少,因此相较于主流的毒化钯碳,本发明的钯碳催化剂可以使用更少的金属钯即可达到更优越的催化效果,催化剂的活性和选择性更高。
18.3、贵金属钯的使用量少,降低了成本。
19.4、使用本发明的钯碳催化剂制得的强力霉素收率高达72%以上。
20.5、本发明使用了噻唑类抑制剂,丰富了抑制剂的选择,对于工业上一些副产物多了一种高效利用的途径,且相较于甲基硫氧嘧啶这种工业上常用的抑制剂,对环境更加友好。
附图说明
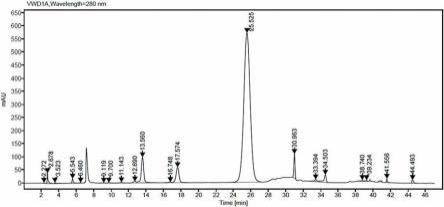
21.图1是本发明实施例5的液相色谱图;图2是本发明实施例6的液相色谱图;图3是本发明实施例7的液相色谱图;图4是本发明实施例8的液相色谱图;图5是本发明对比例1的液相色谱图。
具体实施方式
22.下面结合实施例,进一步阐述本发明。
23.实施例1称取50g杏壳用纯化水清洗表面后用乙醇浸泡2h除去可溶性杂质,120℃烘干至恒重粉碎成颗粒。然后在600℃马弗炉中氮气氛围下炭化1.5h,炭化固体放入浓度为1mol/l的盐酸中预浸渍7h。过滤,并用清水简单冲洗表面后转入质量分数为20%的氢氧化钾溶液中浸渍1h,过滤,其中,炭化固体与盐酸溶液的质量比为1:5,炭化固体与氢氧化钾溶液的质量比为1:5,固体于70℃烘箱中干燥至恒重,在750~850℃马弗炉中氮气氛围下活化2h。活化后固体用80℃的热水打浆4h左右后趁热过滤淋洗,得到活性炭载体a,重量为73.53g,水分含量61.2%。
24.称取折干3.6g的活性炭载体a于500ml高压反应釜四氟内衬中,加入磁力搅拌子后将内衬放到500ml反应釜置于30℃水浴锅中,加入100g纯化水,磁子搅拌转速为100rad/min,持续打浆得活性炭浆液a1。
25.称取4.0000g钯含量为10.00%的氯钯酸溶液用纯化水稀释至80g,然后向内滴入重量为20.56g的乙酸钠溶液,乙酸钠溶液的质量分数为15%,滴加时间为1min,然后在0.9h内缓慢加入质量分数为8.75%的碳酸氢钠溶液,调体系ph至7.25,将该溶液于1.5h内均匀滴入搅拌着的活性炭浆液a1中。然后在30℃水浴锅中继续搅拌12h后调节磁子转速为300rad/min,反应釜内加入质量分数为1%的苯并噻唑乙醇溶液10.16g,反应釜用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在1.2mpa氢气压力下加氢反应5h。抽滤后滤饼用100g水打浆5次,每次30min,过滤后最终得到催化剂a。
26.实施例2称取50g杏壳用纯化水清洗表面后用乙醇浸泡2h除去可溶性杂质,120℃烘干至恒重粉碎成颗粒。然后在600℃马弗炉中氮气氛围下炭化1.5h,炭化固体放入浓度为1mol/l的盐酸中预浸渍7h。过滤,并用清水简单冲洗表面后转入质量分数为20%的氢氧化钾溶液中浸渍1.5h,过滤,其中,炭化固体与盐酸溶液的质量比为1:10,炭化固体与氢氧化钾溶液的质量比为1:12,固体于70℃烘箱中干燥至恒重,在750~850℃马弗炉中氮气氛围下活化2h。活化后固体用82℃的热水打浆5h左右后趁热过滤淋洗,得到活性炭载体b,重量为71.91g,水
分含量60.7%。
27.称取折干3.6g的活性炭载体b于500ml高压反应釜四氟内衬中,加入磁力搅拌子后将内衬放到500ml反应釜置于30℃水浴锅中,加入100g纯化水,磁子搅拌转速为100rad/min,持续打浆得活性炭浆液b1。
28.称取4.0000g钯含量为10.00%的氯钯酸溶液用纯化水稀释至80g,然后向内滴入乙酸钾溶液24.59g,滴加时间为2min,乙酸钾溶液的质量分数为15%,然后在1.1h内缓慢加入质量分数为8.75%的碳酸钠溶液,调体系ph至7.75,将该溶液于2h内均匀滴入搅拌着的活性炭浆液b1中。然后在30℃水浴锅中继续搅拌12h后调节磁子转速为300rad/min,反应釜内加入质量分数为1%的苯并噻唑乙醇溶液10.16g,反应釜用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在1.2mpa氢气压力下加氢反应5h。抽滤后滤饼用100g水打浆5次,每次30min,过滤后最终得到催化剂b。
29.实施例3称取50g竹子用纯化水清洗表面后用乙醇浸泡2h除去可溶性杂质,120℃烘干至恒重粉碎成颗粒。然后在600℃马弗炉中氮气氛围下炭化1.5h,炭化固体放入浓度为1mol/l的盐酸中预浸渍8h。过滤,并用清水简单冲洗表面后转入质量分数为20%的氢氧化钾溶液中浸渍1.8h,过滤,其中,炭化固体与盐酸溶液的质量比为1:18,炭化固体与氢氧化钾溶液的质量比为1:20,固体于70℃烘箱中干燥至恒重,在750~850℃马弗炉中氮气氛围下活化2h。活化后固体用83℃的热水打浆5h左右后趁热过滤淋洗,得到活性炭载体c,重量为73.13g,水分含量61.4%。
30.称取折干3.6g的活性炭载体c于500ml高压反应釜四氟内衬中,加入磁力搅拌子后将内衬放到500ml反应釜置于30℃水浴锅中,加入100g纯化水,磁子搅拌转速为100rad/min,持续打浆得活性炭浆液c1。
31.称取4.0000g钯含量为10.00%的氯钯酸溶液用纯化水稀释至80g,然后向内滴入丙酸钠溶液24.07g,滴加时间为4min,丙酸钠溶液的质量分数为15%,然后在1.1h内缓慢加入质量分数为8.75%的碳酸钠溶液,调体系ph至7.8,将该溶液于2.2h内均匀滴入搅拌着的活性炭浆液c1中。然后在30℃水浴锅中继续搅拌12h后调节磁子转速为300rad/min,反应釜内加入质量分数为1%的苯并噻唑乙醇溶液10.16g,反应釜用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在1.2mpa氢气压力下加氢反应6h。抽滤后滤饼用100g水打浆5次,每次30min,过滤后最终得到催化剂c。
32.实施例4称取50g竹子用纯化水清洗表面后用乙醇浸泡2h除去可溶性杂质,120℃烘干至恒重粉碎成颗粒。然后在600℃马弗炉中氮气氛围下炭化1.5h,炭化固体放入浓度为1mol/l的盐酸中预浸渍9h。过滤,并用清水简单冲洗表面后转入质量分数为20%的氢氧化钾溶液中浸渍2h,过滤,其中,炭化固体与盐酸溶液的质量比为1:25,炭化固体与氢氧化钾溶液的质量比为1:25,固体于70℃烘箱中干燥至恒重,在750~850℃马弗炉中氮气氛围下活化2h。活化后固体用85℃的热水打浆6h左右后趁热过滤淋洗,得到活性炭载体d,重量为72.67g,水分含量61.4%。
33.称取折干3.6g的活性炭载体d于500ml高压反应釜四氟内衬中,加入磁力搅拌子后将内衬放到500ml反应釜置于30℃水浴锅中,加入100g纯化水,磁子搅拌转速为100rad/
min,持续打浆得活性炭浆液d1。
34.称取4.0000g钯含量为10.00%的氯钯酸溶液用纯化水稀释至80g,然后向内滴入顺丁烯二酸二钠20.05g,滴加时间为4min,顺丁烯二酸二钠溶液的质量分数为15%,然后在1.2h内缓慢加入质量分数为8.75%的碳酸氢钠溶液,调体系ph至8,将该溶液于2.5h内均匀滴入搅拌着的活性炭浆液d1中。然后在30℃水浴锅中继续搅拌12h后调节磁子转速为300rad/min,反应釜内加入质量分数为1%的苯并噻唑乙醇溶液10.16g,反应釜用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在1.2mpa氢气压力下加氢反应7h。抽滤后滤饼用100g水打浆5次,每次30min,过滤后最终得到催化剂d。
35.实施例5称取15g的11α-氯-6-次甲基土霉素对甲苯磺酸盐和75g的乙醇,分别放入250ml高压反应釜四氟内衬中,加入折干重量为0.075g的催化剂a和0.75g的纯化水,高压反应釜的磁子搅拌转速设置为300 rad/min,高压反应釜放置于40℃水浴锅中保温搅拌20min,然后用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在0.80mpa氢气压力下加氢反应2h。泄压开釜后称量计算出内衬中料液质量,收率为81.32%。用注射器吸取内衬中料液并经过滤头过滤后取滤液0.2g,用n,n-二甲基甲酰胺定容至10ml后进液相检测,得液相谱图如图1所示。
36.实施例6称取15g的11α-氯-6-次甲基土霉素对甲苯磺酸盐和75g的乙醇,分别放入250ml高压反应釜的四氟内衬中,加入折干重量为0.075g的催化剂b和0.75g的纯化水,高压反应釜的磁子搅拌转速设置为300 rad/min,高压反应釜放置于40℃水浴锅中保温搅拌20min,然后用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在0.80mpa氢气压力下加氢反应2h。泄压开釜后称量计算出内衬中料液质量,收率为81.17%。用注射器吸取内衬中料液并经过滤头过滤后取滤液0.2g,用n,n-二甲基甲酰胺定容至10ml后进液相检测,得液相谱图如图2所示。
37.实施例7称取15g的11α-氯-6-次甲基土霉素对甲苯磺酸盐和75g的乙醇,分别放入250ml高压反应釜的四氟内衬中,加入折干重量为0.075g的催化剂c和0.75g的纯化水,高压反应釜的磁子搅拌转速设置为300 rad/min,高压反应釜放置于40℃水浴锅中保温搅拌20min,然后用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在0.80mpa氢气压力下加氢反应2h。泄压开釜后称量计算出内衬中料液质量,收率为78.27%。用注射器吸取内衬中料液并经过滤头过滤后取滤液0.2g,用n,n-二甲基甲酰胺定容至10ml后进液相检测,得液相谱图如图3所示。
38.实施例8称取15g的11α-氯-6-次甲基土霉素对甲苯磺酸盐和75g的乙醇,分别放入250ml高压反应釜的四氟内衬中,加入折干重量为0.075g的催化剂d和0.75g的纯化水,高压反应釜的磁子搅拌转速设置为300 rad/min,高压反应釜放置于40℃水浴锅中保温搅拌20min,然后用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在0.80mpa氢气压力下加氢反应2h。泄压开釜后称量计算出内衬中料液质量,收率为72.56%。用注射器吸取内衬中料液并经过滤头过滤后取滤液0.2g,用n,n-二甲基甲酰胺定容至10ml后进液相检测,得
液相谱图如图4所示。
39.对比例1称取15g的11α-氯-6-次甲基土霉素对甲苯磺酸盐和75g的乙醇,分别加入至250ml高压反应釜的四氟内衬中,加入折干0.075g的外购新昌公盛材料有限公司生产的,批号为fp2220630的毒化pd/c和0.75g纯化水,磁子搅拌转速设置为300rad/min,高压反应釜放置于40℃水浴锅中保温搅拌20min,然后用0.50mpa氮气置换空气三次,再用0.50mpa氢气置换氮气三次,最后在0.80mpa氢气压力下加氢反应2h。泄压开釜后称量计算出内衬中料液质量,计算收率为70.21%。用注射器吸取内衬中料液并经过滤头过滤后取滤液0.2g,用n,n-二甲基甲酰胺定容至10ml后进液相检测,得液相谱图如图5所示。
40.对实施例5-8和对比例1的数据进行汇总,得下表:应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明讲授的内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。