1.本发明属于生物医药领域,具体涉及一种通便灵胶囊指纹图谱及其构建方法和应用。
背景技术:
2.通便灵胶囊为一种中药成方制剂,具有泻热导滞、润肠通便的作用。用于热结便秘,长期卧床便秘,一时性腹胀便秘,老年习惯性便秘。通便灵胶囊组方为番泻叶、当归、肉苁蓉。番泻叶是一种常用的泻下药,具有泻热行滞、通便、利水之功能,主治热结积滞、便秘腹痛、水肿胀满,已成为全世界范围内应用最广的导泻剂,其主要成分蒽酮及衍生物、黄酮类等。当归具有补血活血、调经止痛、润肠通便的功能,主要化学成分为挥发油、有机酸等。肉苁蓉具有补肾益精、润肠通便,利尿通便、改善记忆力、抗疲劳、抗辐射等作用。
3.目前中药质量标准主要采用药材或者中成药制剂中单一或几个化合物含量进行定性或者定量。中成药成分复杂,仅采用以上定性和定量方式,无法全面和准确评价药品疗效和质量。
4.目前通便灵胶囊质量控制体现在当归药材的薄层鉴别和番泻苷b的含量控制两方面,缺少对肉苁蓉的质量控制,番泻苷b的检测手段为紫外分光光度法,受限于该方法的局限性和番泻苷成分热不稳定性及毒副作用,无法对该产品质量和疗效进行准确控制。研究发现通便灵胶囊中芦荟大黄素等成分也具有泻下的作用,且肉苁蓉的有效成分松果菊苷能改善肠胃功能的作用,仅以单一番泻苷b含量无法全面衡量产品质量。
5.中药指纹图谱技术能较全面的反映中成药所含化学成分的种类和数量,对药品质量进行整体描述和评价。方法开发方便、稳定、有效,对未知和已知成分的分析具有高度特异性和选择性。该方法具有系统系、特征性和稳定性,采用中药指纹图谱技术对中药复杂混合体系的质量进行控制,对于稳定生产工艺、提高药品质量和疗效具有重要作用。现有技术中已经有基于指纹图谱对通便灵胶囊进行质量评价的记载(蔡杨靖,潘云.基于指纹图谱及多指标成分定量的通便灵胶囊质量评价[j].中国药师,2019,22(05):39-42.)。
技术实现要素:
[0006]
本发明所要解决的技术问题是为克服现有技术中缺乏一种尽可能全面的、定性鉴定通便灵胶囊中的活性成分的方法的缺陷,提供一种通便灵胶囊指纹图谱及其构建方法和应用。本发明所提供的指纹图谱对通便灵胶囊药材和多个有效成分进行定性鉴别,节约检测成本,能充分反映产品疗效,从而最终达到控制产品质量的目的,对于通便灵胶囊质量控制具有十分重要的意义。
[0007]
本发明主要通过以下技术方案解决上述技术问题。
[0008]
本发明的技术方案之一为:一种通便灵胶囊指纹图谱的构建方法,所述的构建方法包括以下步骤:利用高效液相色谱检测供试品溶液,将检测结果生成指纹图谱;
[0009]
所述供试品溶液的制备方法包括:将所述通便灵胶囊与60%的甲醇按照(0.4~
0.6)g:50ml的质量体积比混匀;
[0010]
所述高效液相色谱的色谱柱为十八烷基硅烷键合硅胶色谱柱;
[0011]
所述高效液相色谱的色谱条件为:
[0012]
流动相包括流动相a与流动相b,所述流动相a为乙腈,所述流动相b为0.1%磷酸水溶液;
[0013]
梯度洗脱程序:
[0014]
时间(min)0510163040606569乙腈(%)331518202590330.1%磷酸(%)979785828075109797
[0015]
其中,表格中的百分比为流动相a与流动相b分别占流动相总体积的体积百分比。
[0016]
本发明中,所述十八烷基硅烷键合硅胶色谱柱的柱温可为本领域常规,优选20~25℃。
[0017]
本发明中,所述流动相的流速较佳地为1.0ml/min。
[0018]
本发明中,所述高效液相色谱的检测波长可为本领域常规,较佳地为200~260nm,更佳地为220nm。
[0019]
本发明中,所述高效液相色谱的进样体积较佳地为5~20μl;更佳地为10μl。
[0020]
本发明中,所述十八烷基硅烷键合硅胶色谱柱较佳地为ymc-triant c18色谱柱;所述ymc-triant c18色谱柱的规格优选4.6mm
×
250mm,5μm,12nm。
[0021]
在本发明一较佳实施方案中,所述通便灵胶囊与60%的甲醇按照0.5g:50ml的质量体积比混匀。
[0022]
此外,在本发明所述的构建方法中优选使用混合对照品。混合对照品溶液优选包括番泻苷a对照品、番泻苷b对照品、松果菊苷对照品、芦荟大黄素对照品和大黄酸对照品。
[0023]
本发明的技术方案之二为:一种利用如技术方案之一所述的构建方法获得的通便灵胶囊的指纹图谱,所述指纹图谱包括16个峰,且:
[0024]
(1)以6号峰松果菊苷、15号峰芦荟大黄素、16号峰大黄酸为对照峰:
[0025]
1号峰的保留时间为12.363min、rsd为0.151%;
[0026]
2号峰的保留时间为13.969min、rsd为0.104%;
[0027]
3号峰的保留时间为15.870min、rsd为0.125%;
[0028]
4号峰的保留时间为16.890min、rsd为0.135%;
[0029]
5号峰的保留时间为18.427min、rsd为0.175%;
[0030]
6号峰的保留时间为19.868min、rsd为0.201%;
[0031]
7号峰的保留时间为20.797min、rsd为0.179%;
[0032]
8号峰的保留时间为23.175min、rsd为0.210%;
[0033]
9号峰的保留时间为26.738min、rsd为0.215%;
[0034]
10号峰的保留时间为28.241min、rsd为0.239%;
[0035]
11号峰的保留时间为29.489min、rsd为0.250%;
[0036]
12号峰的保留时间为31.190min、rsd为0.231%;
[0037]
13号峰的保留时间为43.030min、rsd为0.144%;
[0038]
14号峰的保留时间为48.695min、rsd为0.029%;
[0039]
15号峰的保留时间为53.920min、rsd为0.019%;
[0040]
16号峰的保留时间为54.537min、rsd为0.018%;
[0041]
或者(2),当以6号峰松果菊苷和16号峰大黄酸为对照峰时:1号峰的相对保留时间0.620~0.625,2号峰的相对保留时间0.700~0.705,3号峰的相对保留时间0.797~0.801,4号峰的相对保留时间0.848~0.852,5号峰的相对保留时间0.926~0.928,7号峰的相对保留时间1.045~1.048,8号峰的相对保留时间1.165~1.168,9号峰的相对保留时间1.344~1.347,10号峰的相对保留时间1.419~1.425,11号峰的相对保留时间1.481~1.488,12号峰的相对保留时间1.566~1.574,13号峰的相对保留时间2.163~2.170,14号峰的相对保留时间2.450~2.451,15号峰的相对保留时间2.714,16号峰的相对保留时间2.745。
[0042]
在本发明一较佳实施方案中,所述指纹图谱为如图4所示的高效液相色谱图。
[0043]
本发明的技术方案之三为:一种通便灵胶囊的质量的检测方法,所述检测方法为根据如技术方案之一所述的构建方法生成供试品的指纹图谱;
[0044]
若所生成的指纹图谱同如技术方案之二所述的指纹图谱,则所述供试品为合格产品。
[0045]
本发明的技术方案之四为:一种记载了如技术方案之二所述的指纹图谱的产品。
[0046]
所述的产品可为本领域的常规形式,例如计算机存储介质;或者,为绘制或印刷的说明书。
[0047]
本发明的技术方案之五为:一种如技术方案之二所述的指纹图谱作为标准指纹图谱在通便灵胶囊检测与质量控制中的应用。
[0048]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0049]
本发明所用试剂和原料均市售可得。
[0050]
本发明的积极进步效果在于:
[0051]
本发明所提供的指纹图谱对通便灵胶囊药材和多个有效成分例如芦荟大黄素、大黄酸、松果菊苷等进行定性鉴别,能充分反映产品疗效,对控制工艺稳定性、保障产品质量、稳定产品疗效具有重要意义;且本发明的构建方法简单、有效、可操作性强,节约检测成本,减少检测时间。
附图说明
[0052]
图1为实施例1中不同样品溶剂分离效果图。
[0053]
图2为实施例1中不同色谱柱温度样品分离效果图。
[0054]
图3为实施例1中不同梯度下样品分离效果图。
[0055]
图4为实施例2中对照指纹图谱。
[0056]
图5为实施例3中批量验证数据所得图谱。
[0057]
图6为实施例3中批量验证样品相似度结果。
具体实施方式
[0058]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商
品说明书选择。
[0059]
以下实施例中所用通便灵胶囊样品均购自武汉康乐药业股份有限公司,国药准字为z20043390。
[0060]
实施例1色谱条件的确定
[0061]
1、样品溶剂选择
[0062]
取通便灵胶囊粉末0.5g,精密称定,置于50ml容量瓶中,加入样品溶剂约40ml,超声使其充分溶解,静置至室温,定容至刻度,摇匀,即得。样品溶剂见表1-1。
[0063]
表1-1样品溶剂表
[0064]
甲醇乙醇20%甲醇20%乙醇60%甲醇60%乙醇
[0065]
其结果如图1所示:60%甲醇从出峰个数到分离情况均优于甲醇、20%甲醇、乙醇、20%乙醇、60%乙醇。故采用60%甲醇为样品溶剂。
[0066]
2、柱温的选择
[0067]
比较色谱柱温度20℃、25℃、30℃下同一样品分离效果,如图2所示:20℃和25℃分离效果均合适,30℃部分样品分离效果较差,因此认为色谱柱温度在20℃和25℃均能达到分离效果。
[0068]
3、波长的选择
[0069]
综合3个目标峰(6号峰松果菊苷、15号峰芦荟大黄素、16号峰大黄酸)的全波长扫描图来看,220nm的波长可同时使得3个目标峰均有较高的吸收。故选择220nm为检测波长。
[0070]
4、梯度方法的优化
[0071]
采用ymc-triant c18(4.6mm
×
250mm,5μm,12nm)色谱柱;流动相为:乙腈-0.1%磷酸水溶液,柱温:25℃,流速:1.0ml/min,检测波长为220nm。各梯度方法及其结果如下:方法梯度3的分离效果最佳,目标峰基本完全分开(图3)。
[0072]
表1-2方法梯度1
[0073]
时间(min)051040606568乙腈%33204090330.1%磷酸(%)97978060109797
[0074]
表2方法梯度2
[0075]
时间(min)05102040606569乙腈%3315202590330.1%磷酸(%)9797858075109797
[0076]
表3方法梯度3
[0077]
时间(min)0510163040606569乙腈%331518202590330.1%磷酸(%)979785828075109797
[0078]
实施例2样品检测情况
[0079]
1.供试品溶液的制备:取通便灵胶囊粉末0.5g,精密称定,置于50ml的容量瓶中,
加入60%甲醇溶液约40ml,摇匀,超声45min,静置至室温,定容至刻度,摇匀即得。
[0080]
2.对照品溶液的制备:取混合对照品,其中松果菊苷4.73mg,番泻苷b6.41mg,番泻苷a 3.96mg,芦荟大黄素1.49mg,大黄酸2.69mg精密称定,置于50ml容量瓶中,加入60%甲醇溶液约40ml,摇匀,超声45min,静置至室温,定容至刻度,摇匀即得。所有对照品番泻苷a和番泻苷b来自stanford chemicals。番泻苷b批号:snd-390952,含量99.35%;番泻苷a批号:snd-390063,含量99.63%。松果菊苷、芦荟大黄素、大黄酸均来自中国食品药品检定研究院。松果菊苷批号:111670-201907,含量91.8%;芦荟大黄素批号:110795-202011,含量97.5%;大黄酸批号:110757-201607,含量99.3%。
[0081]
3.设置高效液相检测条件:采用ymc-triant c18(4.6mm
×
250mm,5μm,12nm)色谱柱;流动相为:乙腈-0.1%磷酸水溶液,柱温:25℃,流速:1.0ml/min,检测波长为220m。理论板数按芦荟大黄素不低于1000000、大黄酸不低于1100000。
[0082]
梯度洗脱程序如以上表3。
[0083]
4.分别精密吸取对照品溶液与供试品溶液各10μl,注入液相色谱仪,记录色谱图。按照上述色谱条件检测后,使用中药色谱指纹图谱相似度评价系统(2012.130723版)软件获得对照指纹图谱,如图4所示。其中指纹图谱峰包括16个特征峰,1号峰的保留时间为12.363min、其rsd=0.151%,2号峰的保留时间为13.969min、其rsd=0.104%,3号峰的保留时间为15.870min、其rsd=0.125%,4号峰的保留时间为16.890min、其rsd=0.135%,5号峰的保留时间为18.427、其rsd=0.175%,6号峰的保留时间为19.868min、其rsd=0.201%,7号峰的保留时间为20.797min、其rsd=0.179%,8号峰的保留时间为23.175min、其rsd=0.210%,9号峰的保留时间为26.738min、其rsd=0.215%,10号峰的保留时间为28.241min、其rsd=0.239%,11号峰的保留时间为29.489min、其rsd=0.250%,12号峰的保留时间为31.190min、其rsd=0.231%,13号峰的保留时间为43.030min、其rsd=0.144%,14号峰的保留时间为48.695min、其rsd=0.029%,15号峰的保留时间为53.92min、其rsd=0.019%,16号峰的保留时间为54.537min、其rsd=0.018%。
[0084]
当以6号峰松果菊苷和16号峰大黄酸为参照峰时,16个特征峰的相对保留时间为:1号峰的相对保留时间0.620~0.625,2号峰的相对保留时间0.700~0.705,3号峰的相对保留时间0.797~0.801,4号峰的相对保留时间0.848~0.852,5号峰的相对保留时间0.926~0.928,7号峰的相对保留时间1.045~1.048,8号峰的相对保留时间1.165~1.168,9号峰的相对保留时间1.344~1.347,10号峰的相对保留时间1.419~1.425,11号峰的相对保留时间1.481~1.488,12号峰的相对保留时间1.566~1.574,13号峰的相对保留时间2.163~2.170,14号峰的相对保留时间2.450~2.451,15号峰的相对保留时间2.714,16号峰的相对保留时间2.745。其中6号峰为松果菊苷、15号峰为芦荟大黄素、16号峰为大黄酸。
[0085]
实施例3 16批验证数据
[0086]
购买武汉康乐药业股份有限公司的16批通便灵胶囊样品,采用以上施例3中的所述的方法进行检测,结果如下图,其中选取包含松果菊苷、芦荟大黄素、大黄酸在内的3个已知目标峰,结合14个未知特征峰,建立对照图谱r(16),结果如图5所示。可以发现16批样品峰分离度良好,峰型及出峰时间均相似。与对照指纹图谱比较并计算其相似度,如图6所示,相似度均在0.99以上,说明本发明方法构建的指纹图谱测定方法可适用于通便灵胶囊的质量控制。
[0087]
实施例4方法验证
[0088]
本实施例中所用对照峰为7号峰(峰面积最大峰)。
[0089]
4.1精密度实验
[0090]
按照实施例2中的方法制备供试品溶液,所制得的供试品溶液,按照实施例2中的色谱条件对供试品溶液连续进样6次检测,考察仪器的稳定性。其相对保留时间和相对峰面积的检测结果见下表。
[0091]
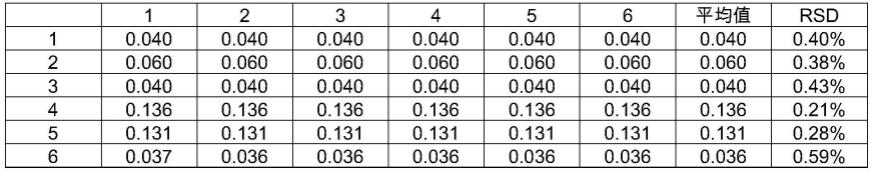
表4通便灵胶囊指纹图谱精密度-相对保留时间
[0092] 123456平均值rsd10.5880.5870.5880.5850.5850.5890.5870.25%20.6660.6650.6660.6640.6640.6660.6650.14%30.7580.7580.7580.7570.7570.7590.7580.14%40.8080.8080.8080.8060.8060.8090.8080.15%50.8790.8790.8790.8770.8760.8800.8780.15%60.9460.9450.9450.9430.9430.9470.9450.15%7(s)1.0001.0001.0001.0001.0001.0001.0000.00%81.1141.1141.1141.1111.1111.1161.1130.20%91.2791.2791.2801.2751.2761.2821.2790.20%101.3611.3601.3621.3561.3571.3641.3600.23%111.4171.4171.4191.4121.4131.4211.4170.24%121.5021.5021.5041.4971.4991.5071.5020.23%132.0402.0382.0422.0372.0392.0432.0400.11%142.3002.2992.3002.2992.3002.3002.3000.02%152.5422.5422.5422.5422.5422.5432.5420.01%162.5712.5712.5722.5712.5722.5722.5710.01%
[0093]
表5通便灵胶囊指纹图谱精密度相对峰面积
[0094][0095][0096]
表4和表5的结果表明,其共有峰的保留时间的rsd值在0.01%~0.25%之间,小于1%,相对峰面积的rsd值在0.21%~1.13%之间,小于5%,符合色谱指纹图谱研究的技术
要求,表明仪器精密度良好。
[0097]
4.2稳定性实验
[0098]
按照实施例2中的方法制备供试品溶液,分别在所制得的供试品溶液后的0、2、4、8、12、24h,按照实施例2中的色谱条件对供试品溶液进行检测,以考察供试品溶液的稳定。其相对保留时间和相对峰面积的检测结果见下表。
[0099]
表6通便灵胶囊指纹图谱稳定性实验-相对保留时间
[0100] 0h2h4h8h12h24h平均值rsd10.5850.5920.5930.5910.5890.5890.5900.52%20.6630.6680.6690.6670.6650.6650.6660.31%30.7560.7600.7610.7590.7570.7580.7590.26%40.8050.8100.8110.8090.8070.8080.8080.27%50.8750.8800.8820.8790.8780.8780.8790.24%60.9410.9450.9480.9450.9440.9450.9450.24%7(s)1.0001.0001.0001.0001.0001.0001.0000.00%81.1081.1141.1191.1141.1121.1131.1130.31%91.2721.2791.2831.2791.2761.2791.2780.30%101.3521.3591.3661.3601.3571.3601.3590.34%111.4071.4151.4221.4161.4131.4171.4150.36%121.4921.5001.5071.5001.4971.5021.5000.35%132.0282.0362.0392.0352.0332.0362.0350.18%142.2922.2932.2942.2932.2922.2932.2930.03%152.5342.5352.5352.5342.5342.5342.5340.02%162.5632.5642.5642.5632.5632.5632.5630.02%
[0101]
表7通便灵胶囊指纹图谱稳定性实验-相对峰面积
[0102][0103][0104]
表6和表7的结果表明,其共有峰的保留时间的rsd值在0.017%~0.516%之间,小于1%,相对峰面积的rsd值在0.56%~2.17%之间,小于5%,符合色谱指纹图谱研究的技术要求,供试品溶液在24h内基本稳定。
[0105]
4.2重复性实验
[0106]
按照实施例2中的方法制备6份供试品溶液,按照实施例2中的色谱条件对供试品溶液进行检测,以考察方法的重复性。其相对保留时间和相对峰面积的检测结果见下表。
[0107]
表8通便灵胶囊指纹图谱重复性实验-相对保留时间
[0108] 123456平均值rsd%10.5950.5950.5950.5950.5950.5960.5950.08%20.6720.6730.6720.6720.6720.6740.6730.09%30.7640.7640.7640.7640.7640.7660.7640.13%40.8120.8120.8120.8120.8130.8150.8130.16%50.8870.8870.8860.8860.8870.8900.8870.16%60.9570.9570.9560.9570.9570.9610.9580.19%7(s)1.0001.0001.0001.0001.0001.0001.0000.00%81.1141.1131.1131.1131.1161.1211.1150.29%91.2871.2851.2861.2851.2881.2941.2880.27%101.3571.3561.3561.3561.3601.3681.3590.34%111.4181.4171.4171.4171.4211.4291.4200.33%121.5001.4991.5001.4991.5031.5121.5020.35%132.0712.0712.0722.0712.0732.0772.0730.12%142.3452.3462.3462.3462.3462.3482.3460.04%152.5972.5982.5982.5982.5982.5992.5980.03%162.6272.6272.6272.6272.6282.6292.6280.03%
[0109]
表9通便灵胶囊指纹图谱稳定性实验-相对保留峰面积
[0110][0111]
[0112]
表8和表9的结果表明,其共有峰的保留时间的rsd值在0.03%~0.35%之间,小于1%,相对峰面积的rsd值在0.30%~1.70%之间,小于5%,符合色谱指纹图谱研究的技术要求,方法的重现性好。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
