1.本发明涉及一种含氰基取代的大环类化合物的晶型及其制备方法,还包括所述晶型在制备治疗ros1基因异常表达的实体瘤的药物中的应用。
背景技术:
2.蛋白激酶在人体内发挥了重要作用,广泛参与了人体内多种细胞的增殖、分化、代谢、凋亡等过程。蛋白激酶的致癌形式在多种不同的人类肿瘤类型中大量表达,并且对一些特定的激酶抑制剂产生高度响应。c-ros原癌基因1受体酪氨酸激酶(c-rosoncogene1receptortyrosinekinase,ros1)属于胰岛素受体超家族,通过调节ras/mapk、pi3k/akt、stat3等主要信号通路,广泛参与细胞生长、增殖和转化,可能参与器官的发育成熟过程。ros1激酶的异常表达,比如基因点突变、过表达和基因融合,均可造成其激酶活性失调,并与许多人类癌症类型相关。
3.ros1融合激酶丢失细胞外区域,保留跨膜和细胞内酪氨酸激酶区域。它不需要配体的结合即可组成性活化,通过磷酸化底物蛋白诱发肿瘤发生及驱动肿瘤细胞的存活和增殖。2007年首次在nsclc病人中发现了cd74-ros1基因融合,迄今为止已有14种以上的伴侣基因被确定。ros1基因融合是继egfr突变、alk融合之后又一明确的nsclc驱动基因,东亚人群阳性发生率约2-3%,欧美约1-2%。2016年3月,仅仅基于50例患者的数据(profile 1001),fda批准克唑替尼(crizotinib)用于治疗ros1基因融合nsclc。克唑替尼疗效显著(客观响应率72%),但约一年后会发展至耐药,其中脑转移(高达47%)和ros1激酶的耐药突变(约50-60%,溶剂前沿突变g2032r约占40%),被证明是克唑替尼主要的耐药机制。对于脑转移的病人,能入脑的药物entrectinib(rxdx-101)和劳拉替尼(lorlatinib)虽可能会带来进一步的pfs获益,但不可避免仍会出现获得性耐药突变。在经历一二线治疗失败后,将面临无药可用。对于获得性g2032r溶剂前沿耐药突变的病人,目前没有上市靶向药物可用。在临床在研ros1抑制剂中,临床实验证实对g2032r溶剂前沿耐药突变病人有效的只有tp therapeutics公司开发的repotrectinib(tpx-0005)。但是和其它所有临床ros1抑制剂一样,repotrectinib(tpx-0005)也是多激酶抑制剂,它有显著的脱靶副作用。特别的是,entrectinib(rxdx-101)和repotrectinib(tpx-0005)都是强的泛ntrk抑制剂,临床上都广泛存在味觉障碍、头晕、感觉异常、体重增加等副作用,这些副作用可能与其对trk激酶的强抑制带来的脱靶相关。对于只有ros1基因融合的病人来说,除了需要承担靶点相关副作用外,还需要额外承担由于脱靶带来的副作用,这将会影响到治疗效果和病人体验。
4.因此,对于ros1基因融合的nsclc的临床治疗,迫切需要一类具有低脱靶副作用的选择性ros1抑制剂、能突破血脑屏障、并对当前市场上药物耐药突变有效的化合物。
5.
技术实现要素:
6.本发明提供了式(i)化合物的a晶型,其特征在于其x射线粉末衍射图谱在下列2θ角处具有特征衍射峰:9.541
±
0.200
°
,18.018
±
0.200
°
,24.120
±
0.20
°
;
[0007][0008]
在本发明的一些方案中,上述a晶型的x射线粉末衍射图谱在下列2θ角处具有特征衍射峰:9.541
±
0.200
°
,18.018
±
0.200
°
,20.839
±
0.200
°
,24.120
±
0.20
°
。
[0009]
在本发明的一些方案中,上述a晶型的x射线粉末衍射图谱在下列2θ角处具有特征衍射峰:9.541
±
0.200
°
,10.758
±
0.200
°
,13.019
±
0.200
°
,18.018
±
0.200
°
,20.839
±
0.200
°
,24.120
±
0.20
°
。
[0010]
在本发明的一些方案中,上述a晶型的x射线粉末衍射图谱在下列2θ角处具有特征衍射峰:9.541
±
0.200
°
,10.758
±
0.200
°
,13.019
±
0.200
°
,15.819
±
0.200
°
,18.018
±
0.200
°
,19.160
±
0.200
°
,20.839
±
0.200
°
,24.120
±
0.20
°
,25.421
±
0.200
°
,28.437
±
0.200
°
。
[0011]
在本发明的一些方案中,上述a晶型的x射线粉末衍射图谱在下列2θ角处具有特征衍射峰:6.661
°
,7.858
°
,9.541
°
,10.758
°
,13.019
°
,13.341
°
,15.819
°
,18.018
°
,18.600
°
,19.160
°
,20.258
°
,20.541
°
,20.839
°
,21.219
°
,21.598
°
,21.961
°
,22.319
°
,22.698
°
,23.021
°
,24.120
°
,24.499
°
,25.421
°
,26.904
°
,27.303
°
,28.122
°
,28.437
°
,28.959
°
,29.662
°
,29.958
°
,30.419
°
,31.457
°
,31.861
°
,32.860
°
,33.698
°
,34.164
°
,34.660
°
,35.556
°
,36.460
°
,37.222
°
,37.738
°
,38.501
°
,39.216
°
。
[0012]
在本发明的一些方案中,上述a晶型,其xrpd图谱如图1所示。
[0013]
本发明的一些方案中,上述a晶型,其xrpd使用cu,kα辐射。
[0014]
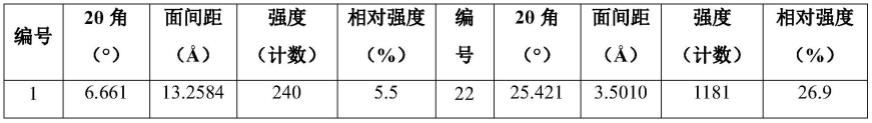
在本发明的一些方案中,上述a晶型的xrpd图谱解析数据如表1所示:
[0015]
表1式(i)化合物a晶型的xrpd图谱解析数据
[0016][0017][0018]
在本发明的一些方案中,上述a晶型的差示扫描量热曲线在259.45
±
3.00℃处具有一个吸热峰的起始点,在302.37
±
3.00℃处具有一个放热峰的峰值。
[0019]
本发明的一些方案中,上述a晶型的dsc分析方法如下:升温速率为10℃/分钟,温度范围为室温~350℃。
[0020]
在本发明的一些方案中,上述a晶型的dsc图谱如图2所示。
[0021]
在本发明的一些方案中,上述a晶型的热重分析曲线在200.0
±
3.0℃时失重达0.097%。
[0022]
在本发明的一些方案中,上述a晶型的tga分析方法如下:升温速率为10℃/分钟,温度范围为室温~500℃。
[0023]
在本发明的一些方案中,上述a晶型的tga图谱如图3所示。
[0024]
本发明还提供了上述a晶型在制备治疗与ros1抑制剂相关病症的药物上的应用。
[0025]
本发明的一些方案中,上述的应用,其特征在于,所述相关药物是用于治疗ros1基因异常表达的实体瘤的药物。
[0026]
本发明的一些方案中,所述实体瘤为非小细胞肺癌。
[0027]
定义和说明
[0028]
除非另有说明,本文所用的下列术语和短语旨在含有下列含义。一个特定的短语或术语在没有特别定义的情况下不应该被认为是不确定的或不清楚的,而应该按照普通的含义去理解。当本文出现商品名时,旨在指代其对应的商品或其活性成分。
[0029]
本发明的中间体化合物可以通过本领域技术人员所熟知的多种合成方法来制备,包括下面列举的具体实施方式、其与其他化学合成方法的结合所形成的实施方式以及本领域技术上人员所熟知的等同替换方式,优选的实施方式包括但不限于本发明的实施例。
[0030]
本发明具体实施方式的化学反应是在合适的溶剂中完成的,所述的溶剂须适合于本发明的化学变化及其所需的试剂和物料。为了获得本发明的化合物,有时需要本领域技术人员在已有实施方式的基础上对合成步骤或者反应流程进行修改或选择。
[0031]
本发明的化合物可以通过本领域技术人员所熟知的常规方法来确认结构,如果本发明涉及化合物的绝对构型,则该绝对构型可以通过本领域常规技术手段予以确证。例如单晶x射线衍射法(sxrd),把培养出的单晶用bruker d8 venture衍射仪收集衍射强度数据,光源为cukα辐射,扫描方式:扫描,收集相关数据后,进一步采用直接法(shelxs97)解析晶体结构,便可以确证绝对构型。
[0032]
下面会通过实施例具体描述本发明,这些实施例并不意味着对本发明的任何限制。
[0033]
本发明所使用的所有溶剂是市售的,无需进一步纯化即可使用。
[0034]
本发明所使用的溶剂可经市售获得。
[0035]
化合物依据本领域常规命名原则或者使用软件命名,市售化合物采用供应商目录名称。
[0036]
技术效果
[0037]
本发明化合物晶型稳定、受热湿度影响小且具有良好的体内给药药效,成药前景广阔;式(i)化合物a晶型具有高选择性ros1激酶的抑制作用;对ros1融合的细胞株展现了较高的细胞增殖抑制活性。
[0038]
本发明x-射线粉末衍射(x-ray powder diffractometer,xrpd)方法
[0039]
仪器型号:dx-2700bh型x射线粉末衍射仪,丹东浩元仪器有限公司
[0040]
测试方法:大约20mg样品用于xrpd检测。
[0041]
详细的xrpd参数如下:
[0042]
射线源:cu,kα
[0043]
光管电压:40kv,光管电流:30ma
[0044]
发散狭缝:1mm
[0045]
主索勒狭缝:28mm
[0046]
二级狭缝:28mm
[0047]
探测器狭缝:0.3mm
[0048]
防散射狭缝:1mm
[0049]
扫描角度范围:3-40deg
[0050]
扫描时间:0.5秒
[0051]
步长:0.02度
[0052]
本发明差热分析(differential scanning calorimeter,dsc)方法
[0053]
仪器型号:mettler toledo dsc1差示扫描量热仪
[0054]
测试方法:取样品(1~10mg)置于dsc铝盘内进行测试,在50ml/min n2条件下,以10℃/min的升温速率,加热样品从室温到350℃。
[0055]
本发明热重分析(thermal gravimetric analyzer,tga)方法
[0056]
仪器型号:mettler tga2 sf/1100热重分析仪
[0057]
测试方法:取样品(2~15mg)置于氧化铝坩埚内进行测试,在50ml/min n2条件下,以10℃/min的升温速率,加热样品从室温到500℃。
[0058]
本发明动态蒸汽吸附分析(dynamic vapor sorption,dvs)方法
[0059]
仪器型号:intrinsic动态蒸汽吸附仪
[0060]
测试方法:取样品(10~30mg)置于tga铝盘内进行测试。
[0061]
详细的dvs参数如下:
[0062]
温度:25℃
[0063]
平衡:dm/dt=0.002%/min(最短:10min,最长:180min)
[0064]
rh(%)测试梯级:10(0-90%),5(90-95%)
[0065]
rh(%)测试梯级范围:0-95-0
[0066]
引湿性评价分类如下:
[0067]
吸湿性分类δw%潮解吸收足量水分形成液体极具吸湿性δw%≥15%有吸湿性15%》δw%≥2%略有吸湿性2%》δw%≥0.2%无或几乎无吸湿性δw%《0.2%
[0068]
注:δw%表示受试品在25
±
1℃和80
±
2%rh下的吸湿增重。
附图说明
[0069]
图1为式(i)化合物a晶型的cu-kα辐射的xrpd谱图;
[0070]
图2为式(i)化合物a晶型的dsc谱图;
[0071]
图3为式(i)化合物a晶型的tga谱图;
[0072]
图4为式(i)化合物a晶型的dvs谱图;
[0073]
图5为式(i)化合物立体结构椭球图;
[0074]
图6为式(i)化合物a晶型对ba/f3 cd74-ros1-wt细胞皮下移植瘤生长的影响。
具体实施方式
[0075]
为了更好的理解本发明的内容,下面结合具体实施例来做进一步的说明,但具体的实施方式并不是对本发明的内容所做的限制。
[0076]
参考例1:中间体a-1的合成
[0077][0078]
步骤1:化合物a-1-2的合成
[0079]
在50l夹套反应釜中,将化合物a-1-1(1500g,7.28mol,1eq)加入到n,n-二甲基甲酰胺(7500ml)和水(3000ml)的混合溶剂中,搅拌。然后依次加入碳酸钾(1.51kg,10.92mol,1.5eq),四丁基醋酸铵(4.39kg,14.56mol,2eq)和化合物3,3-二甲氧基丙烯(2.23kg,21.84mol,3eq),往反应液中鼓氮气30分钟;氮气气氛下一次性加入醋酸钯(22.50g,100.22mmol,1.38%mol),然后氮气鼓泡5分钟,然后加热至70℃反应2小时,然后升温至95℃搅拌反应16hr。将反应液冷却至0-5℃,往反应体系中滴加盐酸(4m,7.1l,3.91eq),控制內温5-20℃,加入时间为30分钟,当有大量固体析出时,测试ph值,此时ph=2-3,在20℃下反应30分钟。往反应体系中慢慢加入碳酸氢钠调节溶液ph为中性(ph=7),往体系中加入甲基叔丁基醚(5l)。搅拌10分钟后,往体系中继续加入甲基叔丁基醚(10l)搅拌10分钟后,分液。水相用甲基叔丁基醚萃取2次,每次10l。三次萃取后,合并有机相,用饱和食盐水洗涤两次,每次7.5l,有机相用无水硫酸钠干燥(1.5kg),过滤,滤液50℃真空浓缩,得到粗品。将本批次粗品与另一平行批次(相同规模投料量)合并打浆纯化。往粗品中加入正庚烷5l,真空浓缩后,得到黄棕色固体。往粗品中加入甲基叔丁基醚和正庚烷的混合溶剂(9l,甲基叔丁基醚:正庚烷体积比为1:5.6)搅拌1小时后过滤,滤饼用正庚烷(1.5l)洗涤,收集滤饼。将母液60℃真空浓缩后,加入甲基叔丁基醚和正庚烷的混合溶剂(1l,甲基叔丁基醚:正庚烷体积比为1:5.6)搅拌1小时后过滤,滤饼用正庚烷(1.5l)洗涤,收集滤饼。将两批次滤饼合并,40-45℃真空干燥5小时,得到粗品。将(r)-二苯基脯氨醇三甲基硅醚(714.78g,2.20mol)溶于二氯甲烷(8000ml)中,降温至0~5℃,在搅拌下加入上述粗品和苯甲酸(268.15g,2.20mol),在0~5℃下搅拌0.5hr,然后在-5~-7℃下加入n-羟基氨基甲酸苄酯(2.20kg,13.17mol),在-5~10℃下搅拌反应16hr。将反应液冷却至0-5℃后,减压过滤,滤饼用二氯甲烷(1500ml)洗涤,滤饼40℃真空干燥16小时,得到白色固体a-1-2。1h nmr(400mhz,cdcl3)δ:7.98(d,j=2.8hz,1h),7.57(dd,j=2.8,8.2hz,1h),7.46-7.37(m,5h),5.86(d,j=4.6hz,1h),5.62(t,j=7.8hz,1h),5.30(q,j=12.3hz,2h),4.02(s,3h),3.01(dd,j=8.5,12.9hz,1h),2.21-2.13(m,1h);lcms m/z=349.2[m h]
。
[0080]
步骤2:化合物a-1-3的合成
[0081]
将a-1-2(542g,1.56mol,1eq)分散于meoh(2600ml)中,将内温降至0℃,分批加入硼氢化钠(82.41g,2.18mol,1.4eq),控制反应温度0-15℃,总加料时间为1.5小时。加毕,在20℃下,继续反应2hr。将本批次与另一平行批次反应液合并后处理(相同规模投料量)。往反应液中加2000ml饱和氯化氨溶液淬灭反应,搅拌5分钟,加入800ml水搅拌后反应液澄清。
反应液用甲基叔丁基醚萃取三次(第一次4l,第二,三次分别为2l),合并有机相,用饱和氯化钠洗涤2次,每次1000ml,有机相用无水硫酸钠干燥,过滤,滤液在45℃真空浓缩后,用乙腈夹带溶剂(每次加入乙腈体积为400ml,然后真空浓缩,重复两次),得到棕色油状物a-1-3。未经纯化直接用于下一步反应。1h nmr(400mhz,cdcl3)δ:7.89(d,j=2.8hz,1h),7.68(br s,1h),7.66(dd,j=2.8,8.4hz,1h),7.42-7.27(m,5h),5.52(dd,j=4.8,10.8hz,1h),5.27-5.07(m,2h),4.01-3.84(m,1h),3.88(s,3h),3.82-3.70(m,1h),2.55(br s,1h),2.29-2.07(m,2h);lcms m/z=351.1[m h]
。
[0082]
步骤3:化合物a-1-4的合成
[0083]
在5l三口瓶中,25℃下,将a-1-3(482g,1.38mol,1eq)溶于无水四氢呋喃(2410ml),向反应液加入三苯基磷(433.03g,1.65mol,1.2eq),在0℃下滴加加入偶氮二甲酸二异丙酯(417.30g,2.06mol,401.25ml,1.5eq),控制内温为0~15℃(滴加时间为1.5小时),在5-25℃下反应16hr。将本批次与其它三批次平行反应合并处理(a-1-3总投料量1951g)。将反应液40-45℃真空浓缩得到粗品,粗品用甲基叔丁基醚(1900ml)和正庚烷(1900ml)的混合溶剂打浆30分钟,过滤除去大部分三苯基氧磷(滤饼),滤饼用甲基叔丁基醚(1000ml)洗涤,滤液真空浓缩得到粗品。将此粗品用硅胶柱层析分离纯化(100-200目,6kg),梯度洗脱,洗脱剂为正庚烷:乙酸乙酯(体积比从20:1慢慢增加至8.3:1,最后保持为5:1),得到棕色油状物a-1-4。1h nmr(400mhz,cdcl3)δ:7.90(d,j=3.0hz,1h),7.50(dd,j=2.4,8.4hz,1h),7.42-7.32(m,5h),5.44(dd,j=5.2,8.6hz,1h),5.31-5.18(m,2h),4.20-4.09(m,1h),3.96(s,3h),3.95-3.83(m,1h),2.97-2.80(m,1h),2.23-2.09(m,1h);lcms m/z=333.2[m h]
。sfc(柱子:chiralpak ad-3,3μm,0.46cm id
×
5cm l;流动相:a(co2)和b(异丙醇,含0.05%二乙胺);梯度:b%=5~40%,2min,保持40%,1.2min,然后5%,0.8min;流速:4ml/min;波长:220nm;压力:1500psi;保留时间:0.875min,手性异构体过量为98.7%。
[0084]
步骤4:化合物a-1的合成
[0085]
在5l三口瓶中,将a-1-4(600g,1.81mol,1eq)溶解在无水二氯甲烷(1200ml)中,氮气保护,将反应液温度降至5℃。然后接好干燥管,尾气吸收装置(吸收液为4m naoh溶液,带缓冲瓶)。将氢溴酸/乙酸溶液(2.66kg,10.83mol,33%纯度,6eq)通过恒压滴液漏斗加入到反应体系中加入时间约为10分钟。加入后恢复至室温25℃,搅拌反应72hr,将本批次与其它三批次平行反应合并处理(a-1-4总投料量1452g)。将反应液用二氯甲烷(2.9l,2v)稀释,在氮气保护下,将反应液减压抽滤,滤饼用二氯甲烷(2l)洗涤一次,甲基叔丁基醚(1l)洗涤一次,滤饼50℃真空干燥4小时,得到粗品。将此粗品用异丙醇(3020ml)室温25℃搅拌打浆1小时,然后减压过滤,滤饼45℃真空干燥得到灰色固体a-1(一分子氢溴酸盐)。1h nmr(400mhz,cd3od)δ:7.88(ddd,j=0.8,3.1,7.9hz,1h),7.64(t,j=3.1hz,1h),5.05(t,j=8.3hz,1h),4.57(dt,j=3.6,7.8hz,1h),4.37(dt,j=6.0,8.5hz,1h),2.97-2.86(m,1h),2.83-2.73(m,1h);lcms m/z=185.0[m h]
。sfc(柱子:chiralpak ad-3,3μm,0.46cm id
×
5cm l;流动相:a(co2)和b(异丙醇,含0.05%二乙胺);梯度:b%=5~40%,5min,然后40~5%,0.5min,最后保持5%,1.5min;流速:2.5ml/min;波长:220nm;压力:1500psi;保留时间:3.611min,手性异构体过量为98.5%。
[0086]
实施例1:式(i)化合物的制备
[0087][0088]
步骤1:化合物1-2的合成
[0089]
在50l反应釜中,将乙酸乙酯(21l)加入到1-1(2180g,14.05mol,1eq)中,加入碳酸钾(97.10g,702.53mmol,0.05eq),在氮气保护下,将反应液降至內温-5℃后,分批加入n-溴代丁二酰亚胺(3kg,2.42mol,1.25eq),控制反应温度在-5~0℃,每次加料70-120g,每次间隔10分钟,加料时间为4.5小时,然后在0℃下,搅拌16小时。向反应液中加入10l水和10l亚硫酸钠水溶液(由无水亚硫酸钠2000g和1800ml水配制而成),继续搅拌5分钟后,內温自然升至15℃,静置1小时,分液(水相为悬浊液,有机相为澄清溶液)。上清液有机相用10l水洗一次,饱和氯化钠水溶液(7.5l)洗一次,水相废弃,有机相待后面合并干燥。将下层水相过滤,滤饼用乙酸乙酯(5l)洗涤。往滤液中加入乙酸乙酯(10l),搅拌分液。水相用乙酸乙酯(10l)萃取一次,合并有机相,有机相用水(10l)洗涤2次,饱和氯化钠水溶液(7.5l)洗一次。将所有有机相合并,用无水硫酸钠(2kg)干燥,过滤,滤饼用乙酸乙酯(2l)洗涤,滤液45℃真空浓缩得到黄棕色固体粗品。将本批次与另一批次平行反应(相同规模投料量)处理后的粗品合并打浆纯化。将粗品用15l甲醇搅拌打浆30分钟,过滤,滤饼废弃。取母液真空浓缩得到黄色粘稠固体粗品。将此粗品用甲基叔丁基醚(4l)搅拌打浆1hr,过滤,滤饼45℃真空浓缩得到黄色固体化合物1-2。1h nmr(400mhz,cdcl3)δ:5.33(br s,2h),4.26(q,j=7.0hz,2h),1.32(t,j=7.2hz,3h);lcms m/z=233.8[m 1]
。
[0090]
步骤2:化合物1-3的合成
[0091]
将1-2(2.6kg,11.11mol,1eq)和磷酸钾(3.54kg,16.6mol,1.5eq)加入到n,n-二甲基甲酰胺(19.5l)中,搅拌5分钟,然后加入3-乙氧基丙烯酸乙酯(1.6kg,11.11mol,1.0eq),在内温101℃下反应16小时。补加3-乙氧基丙烯酸乙酯(320.30g,2.22mol,0.2eq),继续反应16hr。将反应温度降至0~5℃,向其中加入15l纯化水并使其搅拌均匀,然后往体系中慢慢加入3m的hcl溶液(10l)调节ph至3~4,继续搅拌1hr,经减压过滤得黄色固体,滤饼用水充分洗涤三次,每次10l,然后用的甲醇(5l)洗涤一次,所得滤饼经55℃真空干燥48hr后得到黄色固体化合物1-3。1h nmr(400mhz,cdcl3)δ:9.76(br s,1h),7.97(d,j=8.0hz,1h),6.09(d,j=8.0hz,1h),4.34(q,j=7.2hz,2h),1.35(t,j=7.2hz,3h)。
[0092]
步骤3:化合物1-4的合成
[0093]
氮气保护下,向50l反应釜中加入无水n,n-二甲基甲酰胺(15000ml),加毕,开启搅拌,接好安全瓶和次氯酸钠溶液(2.5%)的尾气吸收装置。向反应体系中加入1-3(3.0kg,10.49mol,1eq),搅拌10分钟。氮气保护下,向反应体系中加入氰化亚铜(1.22kg,13.63mol,2.98l,1.3eq)。氮气气氛下,控制体系内温为115-120℃,反应体系保温搅拌48小时。将反应液冷却至约40℃后,将反应液过滤,滤饼用n,n-二甲基甲酰胺(1000ml)洗涤。搅拌下,将滤液分批慢慢倒入到冷水中(30l,5℃),搅拌5分钟。将反应液过滤,滤饼继续用15l水充分洗涤,所得湿的滤饼用甲醇15l搅拌打浆10分钟,过滤得到滤饼,滤饼经55℃真空干燥48hr,得到化合物1-4(3.15kg,粗品)。粗品未经纯化直接用于下一步反应。1h nmr(400mhz,dmso-d6)δ:12.28(br s,1h),8.68(br d,j=8.0hz,1h),6.41(br d,j=8.0hz,1h),4.34(q,j=6.8hz,2h),1.31(t,j=7.0hz,3h)。
[0094]
步骤4:化合物1-5的合成
[0095]
将1-4(190g,613.71mmol,75%含量,1eq)溶于无水二氯甲烷(1900ml)中,然后加入4-二甲氨基吡啶(15.00g,122.74mmol,0.2eq)和三乙胺(186.30g,1.84mol,256.26ml,3eq),搅拌5分钟后,氮气保护,冰盐水浴下降至内温0℃,一次性加入对甲苯磺酰氯(321.75g,1.69mol,2.75eq),加料完毕,然后慢慢自然恢复至室温20℃继续搅拌,总搅拌时间为22小时。将本批次与另一批次平行反应合并后处理(总投料量为390g)。将反应液倒入到2800ml水中,搅拌5分钟,此时上层水相中有固体悬浮物。将上层水相过滤,滤液用二氯甲烷(1l)萃取,分液。将两次萃取的有机相合并,分别用水(1.6l)和饱和食盐水(1.6l)洗涤,有机相用500g无水硫酸钠干燥,过滤,滤液40℃真空浓缩,得到粘稠物,加入1000ml乙腈,搅拌10min,过滤,滤饼用乙腈(800ml)洗涤,滤液真空浓缩得到粗品。将粗品用甲醇(800ml)搅拌打浆5min,过滤,所得滤饼继续用甲醇(200ml)搅拌打浆15min,滤饼40℃真空干燥得到化合物1-5。1h nmr(400mhz,cdcl3)δ:8.68(d,j=7.2hz,1h),8.36(d,j=8.0hz,2h),7.43(d,j=8.0hz,2h),6.87(d,j=7.6hz,1h),4.54(q,j=7.0hz,2h),2.48(s,3h),1.52(t,j=7.2hz,3h);lcms m/z=387.0[m 1]
。
[0096]
步骤5:化合物1-6的合成
[0097]
将化合物a-1(300g,1.13mol,1.33eq)加入到异丙醇(3000ml)中,然后加入n,n-二异丙基乙胺(292.53g,2.26mol,394.25ml,2.66eq),最后加入化合物1-5(327.96g,0.85mol,1eq),在40℃下搅拌2小时。平行投两锅,将两锅反应体系降温至15℃,然后合并过滤,得滤饼固体(540g),母液浓缩干得1.5kg;然后滤饼使用乙酸乙酯(1200ml)机械搅拌打浆1hr,然后过滤,得滤饼固体(361g)和母液(56g);然后将两次母液合并后浓缩干,然后使用二氯甲烷(3000ml)溶解,加入水(3000ml)萃取分液,水相再用二氯甲烷(1500ml)萃取一次,合并有机相,有机相用饱和食盐水(3000ml)洗涤一次,无水硫酸钠干燥,过滤,滤液浓缩后得粗品(630g),然后粗品使用n-甲基吡咯烷酮(450ml)溶解后,缓慢加入到水(4500ml)中,打浆0.5hr,然后过滤,得滤饼固体(330g),滤饼使用乙酸乙酯(660ml)打浆1hr,然后过滤,得滤饼固体(150g),滤饼使用乙酸乙酯(150ml)打浆1hr,然后过滤,得滤饼固体(117g),将两次滤饼(117g)和(361g)合并后,使用乙酸乙酯(478ml)打浆0.5hr,然后过滤,得化合物1-6。1hnmr(400mhz,dmso-d6)δ:11.75(s,1h),9.01(d,j=7.6hz,1h),7.57(s,1h),7.40(s,1h),7.16(d,j=8.0hz,1h),5.57-5.53(m,1h),4.28-4.26(m,1h),4.21-4.16(m,2h),4.00-3.98(m,1h),2.89-2.88(m,1h),2.24-2.20(m,1h),1.24-1.14(m,3h);lcms m/z=399.1[m
1]
。sfc(柱子:chiralpak as-3,3μm,0.46cm id
×
5cm l;流动相:a(co2)和b(meoh,含0.05%异丙胺);梯度:b%=5~50%,3min;流速:3.4ml/min;波长:220nm;压力:1800psi,保留时间:1.04min,手性异构体过量为99.04%。
[0098]
步骤6:化合物1-7的合成
[0099]
将化合物1-6(197g,494.54mmol,1eq)溶于水(492ml)和四氢呋喃(1477ml)的混合溶液中,然后加入一水合氢氧化锂(83.00g,1.98mol,4eq)置换氮气,反应体系由浑浊变成澄清,在25℃搅拌3小时。平行投两锅,将两锅反应体系过滤,滤液与(70g)批次滤液合并,用1m的盐酸溶液(5000毫升)将滤液调节成ph=3~4,析出固体,然后过滤得滤饼固体,然后将滤饼固体使用meoh(9280ml,20v)打浆2hr,然后过滤,得滤饼固体,然后再将滤饼固体使用meoh(4640ml,10v)打浆2hr,然后过滤,得滤饼固体,滤饼真空干燥得到化合物1-7。lcms m/z=371.0[m h]
。sfc(柱子:chiralpak ad-3,3μm,0.46cm id
×
10cm l;流动相:a(co2)和b(etoh,含0.05%异丙胺);梯度:a:b=60:40,4min;流速:4.0ml/min;波长:220nm;压力:1800psi,保留时间:1.73min,手性异构体过量为100%。
[0100]
步骤7:化合物1-8的合成
[0101]
将化合物1-7(200g,540.11mmol,1eq)加入到n,n-二甲基甲酰胺(2000ml)中,然后加入2-(7-氧化苯并三氮唑)-n,n,n’,n
’‑
四甲基脲六氟磷酸酯(205.37g,540.11mmol,1.0eq)和1-氨基环丙烷甲醇盐酸盐(66.75g,540.11mmol,1.0eq,hcl),置换氮气后,加入n,n-二异丙基乙胺(279.22g,2.16mol,376.31ml,4eq),反应体系由白色浑浊变成黄色澄清再变成浅黄色浑浊,在15~25℃下搅拌1小时。向反应体系中加入饱和氯化氨水溶液(20ml,0.1v),再加入乙腈(800ml,4v)搅拌30分钟后,通过布氏漏斗过滤,滤饼固体与(28g)批次合并后使用乙腈(800ml,4v),在20℃下打浆搅拌30分钟,然后将体系过滤,得滤饼固体。收集滤饼,滤饼真空干燥得到类白色固体化合物1-8。1hnmr(400mhz,dmso-d6)δ:11.86(s,1h),8.98(d,j=7.6hz,1h),7.70(s,1h),7.60(s,1h),7.46-7.43(m,1h),7.11(d,j=8.0hz,1h),5.42(t,j=6.8,1h),4.59-4.58(m,1h),4.34-4.30(m,1h),4.08-4.03(m,1h),3.47-3.38(m,2h),3.03
–
2.95(m,1h),2.25-2.18(m,1h),0.74-0.68(m,2h),0.60-0.57(m,1h),0.35-0.32(m,1h);lcms m/z=440.1[m h]
。sfc(柱子:(s,s)-whelk-o1,3.5μm,0.46cm id
×
5cm l;流动相:a(co2)和b(etoh,含0.05%异丙胺);梯度:b%=5~50%,3min;流速:3.4ml/min;波长:220nm;柱温:35℃;背压:1800psi,保留时间:1.65min,手性异构体过量为99.70%。
[0102]
步骤8:式(i)化合物的合成
[0103]
将化合物1-8(102g,232.14mmol,1eq)溶于四氢呋喃(1020ml)中,置换氮气后,加入三正丁基膦(93.93g,464.27mmol,114.55ml,2eq),再加入偶氮二甲酰二哌啶(117.14g,464.27mmol,2eq),在15~25℃下反应3小时。平行投两锅,向体系中加入meoh(408ml,4v),搅拌30min后,将两锅体系合并过滤得滤饼固体155g,然后将滤饼固体与(20g)批次的固体合并后用meoh(1120ml,5v)打浆,搅拌30min后,过滤,收集滤饼,滤饼真空干燥得到式(i)化合物。1hnmr(400mhz,dmso-d6)δ:9.26(s,1h),8.98(d,j=8.0hz,1h),8.16(d,j=2.8hz,1h),7.89(dd,j=8.4hz,2.8hz,1h),7.04(d,j=8.0hz,1h),5.97(t,j=8.4,1h),4.76(d,j=10.8hz,1h),4.59(t,j=7.2hz,1h),4.02
–
3.99(m,1h),3.91(d,j=10.8hz,1h),3.07-3.01(m,1h),2.63-2.57(m,1h),2.13-2.08(m,1h),1.28-1.24(m,1h),0.98-0.89(m,2h);
lcms m/z=422.1[m h]
。sfc(柱子:chiralcel od-3,3μm,0.46cm id
×
5cm l;流动相:a(co2)和b(meoh,含0.05%异丙胺);梯度:b%=10~40%,3min;流速:4.0ml/min;波长:220nm;压力:100bar,保留时间:2.03min,手性异构体过量为100%。
[0104]
实施例2:式(i)化合物a晶型的制备
[0105]
将式(i)化合物(128g,303.76mmol)加入到二甲基亚砜(1280ml)中,加热至65℃使溶液变为澄清后,过滤,将冷却后的滤液滴加加入到水(3840ml)中,在20℃下搅拌30分钟,过滤,滤饼用甲醇(2560ml)搅拌打浆1小时,过滤,收集滤饼,滤饼在45~55℃下真空干燥得到得到式(i)化合物a晶型。
[0106]
实施例3:式(i)化合物a晶型固体稳定性试验
[0107]
分别称取化合物约20mg置于干燥洁净的玻璃瓶中,称4份,摊成薄薄一层,作为供试样品,放置于影响因素试验条件下(60℃,25℃/92.5%rh)和加速条件下(40℃/75%rh,60℃/75%rh),其样品为完全暴露样。60℃,25℃/92.5%rh在5天、10天取样分析;40℃/75%rh和60℃/75%rh在1个月、2个月、3个月取样分析。
[0108]
表2式(i)化合物a晶型样品含量和有关物质结果(5天、10天、1月、2月、3月)
[0109][0110][0111]
结论:式(i)化合物a晶型在高温、高湿条件下均具有良好的稳定性。
[0112]
实施例4:式(i)化合物a晶型的吸湿性研究
[0113]
实验材料:
[0114]
intrinsic动态蒸汽吸附仪
[0115]
实验方法:
[0116]
取式(i)化合物a晶型10~30mg置于dvs样品盘内进行测试。
[0117]
实验结果:
[0118]
式(i)化合物a晶型的dvs谱图如图4所示,δw=0.1004%。
[0119]
实验结论:
[0120]
式(i)化合物a晶型在25℃和80%rh下的吸湿增重为0.1004%,几乎无引湿性。
[0121]
实施例5:式(i)化合物a晶型单晶x射线衍射检测分析
[0122]
单晶培养过程:取样品式(i)化合物a晶型(约104mg),置于250ml棕色锥形瓶中,使用丙酮(15ml)充分溶解后,室温静置三天后,出现小块状无色晶体。收集晶体,用rigaku oxford diffraction xtalab synergy衍射仪收集衍射强度数据,光源为cukα辐射,结构解析使用shelxt(sheldrick,g.m.2015.actacryst.a71,3-8)。通过式(i)化合物的单晶数据,可以确定其绝对构型。式(i)化合物立体结构椭球图见附图5;式(i)化合物晶体结构数据和参数见表3、4、5、6、7和8。
[0123]
仪器参数与数据收集
[0124]
理学牛津衍射xtalab synergy-s型号四元单晶衍射仪(配备hypix-6000he面检测器);
[0125]
低温系统:oxford cryostream 800;
[0126]
光源:50w,多层反射微焦斑光管;
[0127]
光管电压:50kv;
[0128]
光管电流:1ma;
[0129]
晶体至检测器距离:d=35mm。
[0130]
表3:x射线晶体学数据汇总
[0131][0132]
表4:原子坐标(
×
104)和等价各向同性移位参数
[0133][0134]
表5:键长
[0135][0136]
表6:键角[deg]
[0137]
[0138][0139]
表7:氢键[a和deg]
[0140][0141]
表8:扭转角度[deg]
[0142]
[0143][0144]
生物测试数据
[0145]
实验例1:式(i)化合物a晶型对ros1、trka和alk的激酶抑制活性
[0146]
1.实验药物
[0147]
阳性对照药crizotinib购自江苏倍达医药科技有限公司,entrectinib购自武汉永璨生物科技有限公司,lorlatinib购自购自上海升德医药科技有限公司,repotrectinib购自陕西新研博美生物科技有限公司。
[0148]
2.实验方法
[0149]
式(i)化合物a晶型对ros1、trka和alk的激酶抑制活性测试在reaction biology corp.公司完成。在反应缓冲液(20mm羟乙基哌嗪乙硫磺酸(hepes)(ph 7.5),10mm氯化镁
(mgcl2),1mm乙二醇双氨乙基醚四乙酸(egta),0.02%聚氧乙烯十二烷醚(brij35),0.02mg/ml bsa,0.1mm钒酸钠(na3vo4),2mm二硫苏糖醇(dtt),1%dmso)中依次加入一定浓度的底物、辅酶因子、激酶和测试化合物(10个剂量,3倍连续稀释液,2%dmso最终浓度)并混匀,将混合物在室温下孵育20分钟,向反应混合液中加入一定浓度的
33
p-atp开始反应,随后室温孵育120分钟。最后通过过滤-结合的方法来检测反应物的放射性。最终的激酶活性以测试样品中剩余的激酶活性占dmso对照组的激酶活性的比例来表示。通过graphpad软件拟合量效关系曲线并计算ic
50
。结果如表9:
[0150]
3.实验结果
[0151]
表9:激酶半数抑制浓度ic
50
(nm)
[0152][0153]
结果表明:式(i)化合物a晶型对ros1抑制活性与crizotinib、entrectinib、lorlatinib和repotrectinib基本相当。同时式(i)化合物a晶型对突变激酶ros1-g2032r展现了高度的抑制,抑制活性显著高于crizotinib、entrectinib和lorlatinib,与repotrectinib相当。但是式(i)化合物a晶型对trka和alk激酶抑制活性较弱,其对alk激酶的选择性显著优于crizotinib、entrectinib、lorltinib和repotrectinib,对trka激酶的选择性显著优于entrectinib和repotrectinib。
[0154]
实验例2:式(i)化合物a晶型对细胞增殖的抑制活性
[0155]
三磷酸腺苷(adenosine tri-phosphate,atp)是自然界中各种生命活动中共用的能量载体,是能量储存和转移的最小单位。celltiter-glo
tm
活细胞检测试剂盒采用萤光素酶作检测物,发光过程中萤光素酶需要atp的参与。向细胞培养基中加入celltiter-glo
tm
试剂,测量发光值,光信号和体系中atp量成正比,而atp又和活细胞数正相关。因此通过使用celltiter-glo试剂盒检测atp含量,可以检测出细胞的增殖情况。本测试中,细胞系为ba/f3 slc34a2-ros1-wt、ba/f3 slc34a2-ros1-g2032r、ba/f3 lmna-ntrk1、ba/f3 cd74-ros1、ba/f3 cd74-ros1-g2032r和ba/f3 cd74-ros1-d2033n稳转细胞株(ba/f3 slc34a2-ros1-wt、ba/f3 slc34a2-ros1-g2032r、ba/f3 lmna-ntrk1测试是在康源博创生物科技(北京)有限公司进行测试;ba/f3 cd74-ros1、ba/f3 cd74-ros1-g2032r和ba/f3 cd74-ros1-d2033n在合肥中科普瑞昇生物医药科技有限公司进行测试)。
[0156]
ic
50
测定过程:
[0157]
1细胞培养和接种
[0158]
a)收获处于对数生长期的细胞并采用血小板计数器进行细胞计数。用台盼蓝排斥法检测细胞活力,确保细胞活力在90%以上。
[0159]
b)调整细胞浓度;分别添加90μl细胞悬液至96孔板中。
[0160]
c)将96孔板中的细胞置于37℃、5%co2、95%湿度条件下培养过夜。
[0161]
2药物稀释和加药
[0162]
a)配制10倍药物溶液,最高浓度为10μm,9个浓度,3倍稀释,在接种有细胞的96孔板中每孔加入10μl药物溶液,每个药物浓度设置三个复孔。
[0163]
b)将已加药的96孔板中的细胞置于37℃、5%co2、95%湿度条件下继续培养72小时,之后进行ctg分析。
[0164]
3终点读板
[0165]
a)融化ctg试剂并平衡细胞板至室温30分钟。
[0166]
b)每孔加入等体积的ctg溶液。
[0167]
c)在定轨摇床上振动5分钟使细胞裂解。
[0168]
d)将细胞板放置于室温20分钟以稳定冷光信号。
[0169]
e)读取冷光值。
[0170]
4数据处理
[0171]
使用graphpad prism 5.0软件分析数据,利用非线性s曲线回归来拟合数据得出剂量-效应曲线,并由此计算ic
50
值,数据见表10。
[0172]
表10细胞半数抑制浓度ic
50
(nm)
[0173][0174]
结果表明:式(i)化合物a晶型对ros1融合的细胞株ba/f3 slc34a2-ros1、ba/f3 cd74-ros1及其突变细胞株ba/f3 slc34a2-ros1-g2032r、ba/cd74-ros1-g2032r和ba/f3 cd74-ros1-d2033n展现了较高的细胞增殖抑制活性,且抑制活性显著强于crizotinib和entrectinib,而与repotrectinib相当或优于repotrectinib。
[0175]
实验例3:式(i)化合物a晶型在小鼠体内的药效测试
[0176]
实验目的:评价式(i)化合物a晶型等受试药在ba/f3 cd74-ros1细胞皮下移植肿瘤balb/c裸小鼠模型的体内药效。
[0177]
1实验材料
[0178]
a)实验动物
[0179]
品系:balb/c裸小鼠
[0180]
到货周龄:6-8周龄
[0181]
性别:雌性
[0182]
供应商:北京维通利华实验动物技术有限公司(浙江分公司)
[0183]
b)饲养环境
[0184]
动物在spf级动物房以ivc(独立送风系统,恒温恒湿)笼具饲养(每笼3-4只)
[0185]
温度:20-26℃
[0186]
湿度:40-70%
[0187]
笼具:以聚碳酸酯制成,体积325mm
×
210mm
×
180mm。垫料为玉米芯,每周更换两次。
[0188]
食物:实验动物在整个实验阶段中可自由进食(照射灭菌,干颗粒状食物)。
[0189]
饮水:实验动物可自由饮用灭菌水。
[0190]
笼具标识:每笼动物信息卡应注明笼内动物数目,性别,品系,接收日期,给药方案,实验编号,组别以及实验开始日期。
[0191]
动物标识:实验动物以耳标进行标识。
[0192]
c)肿瘤组织或细胞信息
[0193]
细胞:ba/f3 cd74-ros1-wt细胞体外单层培养,培养条件为rpmi1640培养基中加10%热灭活胎牛血清,37℃5%co2培养。一周两次用胰酶-edta进行常规消化处理传代。当细胞呈指数生长期时,收取细胞,计数,接种。
[0194]
d)化合物信息
[0195]
crizotinib购自上海毕得医药科技有限公司。
[0196]
e)肿瘤组织或细胞信息
[0197]
细胞:ba/f3 cd74-ros1-wt细胞体外单层培养,培养条件为rpmi1640培养基中加10%热灭活胎牛血清,37℃5%co2培养。一周两次用胰酶-edta进行常规消化处理传代。当细胞呈指数生长期时,收取细胞,计数,接种。
[0198]
f)其它试剂信息
[0199]
试剂信息
[0200]
名称生产厂家胎牛血清hyclonerpmi1640培养基gibco胰酶gibco
[0201]
g)仪器信息
[0202]
仪器信息
[0203]
名称生产厂家型号二氧化碳培养箱赛莫飞世尔(thermo fisher)heracell240i低温高速离心机艾本德(eppendorf)5810r分析天平赛多利斯(sartorius)secura225d-1cn普通天平常州天之平仪器设备有限公司el-2kj数显游标卡尺三丰0~150mm
[0204]
2实验方法与步骤
[0205]
a)细胞培养准备
[0206]
ba/f3 cd74-ros1-wt细胞体外培养,rpmi1640培养基中加10%热灭活胎牛血清,37℃5%co2孵箱培养。一周两次用胰酶-edta进行常规消化处理传代。当细胞饱和度为80%-90%,数量到达要求时,收取细胞,计数,接种。
[0207]
b)肿瘤细胞接种及分组
[0208]
细胞接种:将0.2ml(5
×
106个)ba/f3 cd74-ros1-wt细胞皮下接种于每只小鼠的右后背,肿瘤平均体积达到96mm3时开始分组给药。
[0209]
c)药物配制和给药
[0210]
化合物均以10%dmso 10%solutol 80%水为溶媒配成澄清溶液,用于po(灌胃)组给药。给药体积参数:根据小鼠体重10μl/g。如果体重下降超过15%,则停药,直到体重恢复到10%以内再给药。
[0211]
d)肿瘤测量
[0212]
每周两次用游标卡尺测量肿瘤直径。肿瘤体积的计算公式为:v=0.5
×a×
b2,a和b分别表示肿瘤的长径和短径。化合物的抑瘤疗效用tgi(%)评价。tgi(%),反映肿瘤生长抑制率。tgi(%)=[(1-(某处理组给药结束时平均瘤体积-该处理组开始给药时平均瘤体积))/(溶剂对照组治疗结束时平均瘤体积-溶剂对照组开始治疗时平均瘤体积)]
×
100%。结果见图6。
[0213]
e)统计分析
[0214]
统计分析基于试验结束时相对肿瘤体积和肿瘤重量运用spss软件进行分析。多组间比较用one-way anova进行分析,如果方差齐(f值无显著性差异),应用tukey’s法进行分析,如果方差不齐(f值有显著性差异),应用games-howell法进行检验。p《0.05认为有显著性差异。
[0215]
3.实验结果:在ba/f3 cd74-ros1皮下异种移植瘤上给药13天时,式(i)化合物a晶型在低至1.5mg/kg/bid的给药剂量下即具有显著抗肿瘤作用(tgi=83.5%),并且抗肿瘤作用具有量效依赖的趋势。式(i)化合物a晶型在5mg/kg/bid剂量下展现缩瘤(tgi=101.4%),药效等同于30mg/kg/qd剂量下crizotinib的药效(tgi=101.9%)。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
