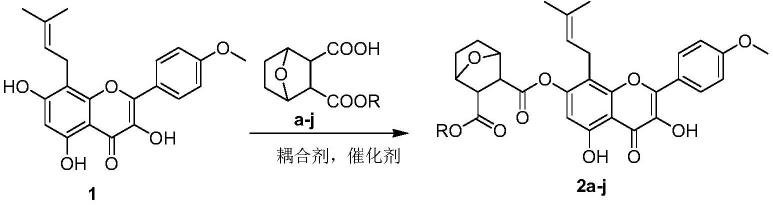
1.本发明属于电化学技术领域,具体来说是新型金属卟啉化合物及其制备方法和在电催化氧气还原中的应用。
背景技术:
2.氧气还原反应在生物和人工能源转换体系中都是非常重要的化学反应过程,在自然体系中,生物体内的细胞进行有氧呼吸时,细胞色素氧化酶(cytochromec oxidase,ccos)会催化氧气还原得到水,同时产生供机体各种生命活动需要的能量(atp);在人工能源转换体系中,燃料电池和金属空气电池是利用氧气还原作为阴极反应的下一代重要能源技术,它们通常结合燃料(如h2)的氧化和氧气还原来产生电能,为人们的日常生活提供动力。
3.可见氧气还原反应是能源转换中重要的化学反应过程,然而氧气还原过程中涉及多个电子和质子的转移,导致其反应能垒高、动力学过程缓慢,不同的电子和质子数目、顺序均会导致不同的选择性,而这些不同的反应路径和选择性是由催化剂来决定的,因此开发高效的氧气还原催化剂,具有重要的科学意义和应用前景。
4.分子催化剂的研究有利于了解反应机理、研究催化剂构效关系;传统氧气还原金属配合物电催化剂存在分子聚集的缺陷,容易引起双分子还原机制,导致不利于催化剂进行选择性调控、均相体系的分子设计很难在多相催化系统中重现的问题,基于此,开发一种全新高效的非贵金属催化剂是当前和未来电催化氧气还原研究的核心问题。
技术实现要素:
5.针对上述现有技术的不足,本发明的目的是提供新型金属卟啉化合物及其制备方法和在电催化氧气还原中的应用,本发明首次提出了构筑含有拱门型位阻基团的新型廉价金属卟啉氧气还原催化剂,并在位阻内引入功能型的吡啶基团,通过拱门型位阻稳定氧气还原的中间体以限制双分子氧还原机制;通过引入分子内质子和电荷促进氧氧键断裂,通过设计合成金属卟啉的刚性位阻结构增强催化剂的单分子特性,进一步实现对均相和多相体系下的构效关系的研究。
6.为解决上述技术问题,本发明采用如下技术方案:
7.一种新型金属卟啉化合物,所述新型金属卟啉化合物的结构如式(i)所示:
[0008][0009]
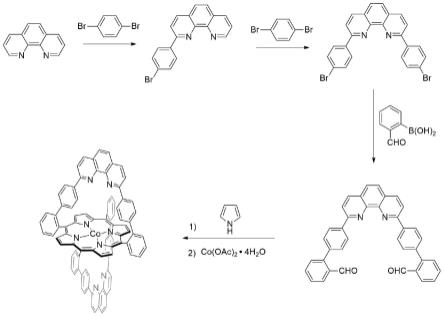
本发明还保护了新型金属卟啉化合物的制备方法,包括如下步骤:
[0010]
(1)将1,10-菲罗啉和对二溴苯加入至乙醚中,于催化条件下进行亲电取代反应,然后经水洗、柱层析纯化,得到化合物1;
[0011]
(2)将步骤(1)的化合物1和对二溴苯加入至乙醚中,于催化条件下进行亲电取代反应,然后经水洗、柱层析纯化,得到化合物2;
[0012]
(3)惰性气氛中,将步骤(2)的化合物2、2-醛基苯硼酸和碱加入至甲醇溶液中,于催化条件下,60-80℃回流10-12h后,用含nh4oh的na2co3溶液提取后,经水洗得到邻菲啰啉,再将邻菲啰啉与二氧化锰进行氧化反应后,柱层析纯化,得到化合物3;
[0013]
(4)于惰性气氛中,将步骤(3)的化合物3和吡咯加入至二氯甲烷中,然后加入三氟乙酸,室温下搅拌18-24h后,向其中加入2,3-二氯-5,6-双氰诺-1,4-苯醌,避光回流2-3h,再经三乙胺中和、萃取、干燥有机相、柱层析纯化,得到化合物4;
[0014]
(5)将步骤(4)的化合物4和醋酸钴盐加入至溶液中,回流反应20-24h后,经水洗、柱层析纯化,得到新型金属卟啉化合物。
[0015]
优选的,所述步骤(1)中1,10-菲罗啉和对二溴苯的物质的量之比为1:1-3,所述步骤(2)中化合物1和对二溴苯的物质的量之比为1:1-5。
[0016]
优选的,所述步骤(1)和所述步骤(2)催化条件的催化剂均选自正丁基锂;所述步骤(1)和所述步骤(2)亲电取代反应的条件均为:于-5-0℃下反应2-2.5h。
[0017]
优选的,所述步骤(3)的邻菲啰啉与二氧化锰反应条件为:将邻菲啰啉和二氧化锰混合于二氯甲烷中,于室温下搅拌反应30-40min;
[0018]
其中,二氧化锰与邻菲啰啉的物质的量之比15-20:1。
[0019]
所述步骤(3)中,化合物2与2-醛基苯硼酸的物质的量之比为1:1-5,所述碱为碳酸铯或碳酸钠,催化条件的催化剂选自四(三苯基膦)钯或者二氯二三苯基磷钯。
[0020]
优选的,所述步骤(4)中,化合物3、吡咯、三氟乙酸、2,3-二氯-5,6-双氰诺-1,4-苯醌的物质的量之比为1:2-3:0.01:1-1.5。
[0021]
优选的,所述步骤(5)中,化合物4和醋酸钴盐的物质的量之比为1:5-10,所述溶液由甲醇和二氯甲烷组成,二者的体积比为1:1-3。
[0022]
本发明还保护了新型金属卟啉化合物在制备电催化氧气还原分子催化剂中的应用。
[0023]
优选的,所述应用的方法为:将新型金属卟啉化合物与碳黑混合后,分散在乙醇中得到浆料,然后将浆料涂覆于电极表面并晾干,再进行氧气还原测试;
[0024]
其中,催化剂与碳黑的质量比为1:1-2。
[0025]
与现有技术相比,本发明具有的有益效果是:
[0026]
1、本技术提出了构筑含有拱门型位阻基团的新型廉价金属卟啉氧气还原催化剂,并在位阻内引入功能型的吡啶基团,以通过拱门型位阻稳定氧气还原的中间体,继而限制双分子氧还原机制;
[0027]
拱门内邻菲啰啉的吡啶n在酸性条件下可以被质子化,就可以提供分子内质子和电子,通过引入分子内质子和电荷促进氧氧键断裂;通过设计合成金属卟啉的刚性位阻结构增强催化剂的单分子特性,进一步实现均相和多相体系下的构效关系研究。
[0028]
2、本发明致力于研究金属卟啉催化剂结构与其电催化氧气还原机制之间的关系,揭示分子化合物电催化氧气还原性能的关键制约因素,最终发展一类高效电催化氧气还原新体系。
[0029]
3、氧气还原反应包括:两个电子还原过程到过氧化氢,四个电子还原过程到水,这也是目前氧气还原反应普遍存在的选择性不高的原因所在;本技术的创新之处在于:该催化剂在碱性条件下通过位阻效应可以实现完全的两电子氧气还原过程,酸性条件下则可以实现四电子氧气还原过程。现有在电催化氧还原研究方面经常难以进行准确的四电子或两电子还原过程,本技术设计的拱门结构催化剂能够准确的完成氧还原的选择性还原。
附图说明
[0030]
图1为本发明实施例1制得的新型金属卟啉化合物的合成路线图;
[0031]
图2为本发明实施例1制得的苯基卟啉的orr转移电子数计算图;其中,(a)为旋转圆盘电极电极测试得到的盘电极电流(下)和环电极电流(上);(b)为根据盘电极电流和环电极电流计算得到的氧气还原电子数;
[0032]
图3为本发明实施例1制得的位阻型卟啉的orr转移电子数计算图,其中,(a)为旋转圆盘电极电极测试得到的盘电极电流(下)和环电极电流(上);(b)为根据盘电极电流和环电极电流计算得到的氧气还原电子数;
[0033]
图4为本发明实施例1制得的双拱门卟啉配体的氢谱图。
具体实施方式
[0034]
下面对本发明的具体实施方式进行详细描述,但应当理解本发明的保护范围并不受具体实施方式的限制。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。本发明各实施例中所述实验方法,如无特殊说明,均为常规方法。
[0035]
实施例1
[0036]
新型金属卟啉化合物的制备方法,包括如下步骤:
[0037]
(1)2-(对溴苯)-1,10-菲罗啉的合成:
[0038]
将1,10-菲罗啉和对二溴苯在乙醚溶液里混合,二者物质的量之比为1:2,加入正丁基锂作为催化剂进行催化,正丁基锂与反应物总物质的量之比为1.3:1,在室温下反应10h,停止反应后,水洗后过柱纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2-(对溴苯)-1,10-菲罗啉;1h nmr(400mhz,cdcl3):9.23(dd,1h),8.32-8.18(m,4h),8.05(d,1h),7.78(d,2h),7.65(dd,2h)(figure s1).hrms:calcd for c18h12n2br[m h] ,335.0178;found,335.0176(figure s2c).anal.calcd.for c18h11n2br:c,64.50;h,3.31;n,8.36.found:c,64.63;h,3.52;n,8.51.
[0039]
(2)2,9-双(对溴苯)-1,10-菲罗啉的合成:
[0040]
将步骤(1)制备的2-(对溴苯)-1,10-菲罗啉和对二溴苯在乙醚溶液中混合,二者物质的量之比为1:3,加入正丁基锂在室温下反应10h,正丁基锂与反应物总物质的量之比为1.3:1,然后进行纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对溴苯)-1,10-菲罗啉;1hnmr(400mhz,cdcl3):8.32(dd,6h),8.13(d,2h),7.81(s,2h),7.72(d,4h)(figure s3).,hrms:calcd for c
24h15
n2br2[m h]
,490.9577;found,490.9569(figure s4c).anal.calcd.for c
24h14
n2br2:c,58.81;h,2.88;n,5.71.found:c,59.05;h,2.93;n,5.79.
[0041]
(3)2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的合成:
[0042]
将步骤(2)制备的2,9-双(对溴苯)-1,10-菲罗啉和2-醛基苯硼酸在甲醇溶液中混合,二者物质的量之比为1:3,加入过量四三苯基膦二氯化钯作为催化剂,四三苯基膦二氯化钯与反应物总物质的量之比为3:1,加入碳酸钠作为碱,碳酸钠与2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的物质的量之比为2:1,在75℃下回流10h后,用含高浓度nh4oh(10ml)的na2co3(2mol/l,100ml)提取两次,反应结束后水洗,然后与二氧化锰共同加入至二氯甲烷中,室温反应35min后,进行过柱处理纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉;1hnmr(400mhz,(cd3)2so):10.04(s,2h),8.68(dd,6h),8.53(d,2h),8.07(s,2h),7.98(d,2h),7.81(t,2h)7.72(d,4h),7.65(dd,4h)(figure s5).hrms:calcd for c
38h24
n2o2na[m na]
,563.1717;found,563.1730(figure s6c).anal.calcd.for c
38h24
n2o2:c,84.42;h,4.47;n,5.18.found:c,84.65;h,4.55;n,5.37.
[0043]
(4)双拱门卟啉的合成:
[0044]
在装有磁性搅拌棒的烧瓶中,加入2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉和吡咯,二者物质的量之比为1:2.5,然后在n2下加入dcm(400ml),然后加入三氟乙酸,三氟乙酸与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉的物质的量之比为0.01:1,在室温下搅拌20h后,加入2,3-二氯-5,6-双氰诺-1,4-苯醌,2,3-二氯-5,6-双氰诺-1,4-苯醌与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉等物质的量,避光回流2h,然后用三乙胺中和,加水三次萃取后,用mgso4干燥有机相,然后使用柱层析进行纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门卟啉;1hnmr(400mhz,cdcl3):8.76(d,2h),8.69-8.54(m,6h),8.37(td,2h),8.16(d,1h),8.08(s,1h),8.06(dd,2h),7.99(d,2h),7.85-7.78(m,6h),7.75-7.60(m,7h),7.59-7.51(m,7h),7.08(td,4h),6.91(d,4h),6.84(d,2h),6.64(d,6h),-2.92(s,2h)(figure s7).hrms:calcd for c
92h55
n8[m h] ,1271.4544;found,1271.4542(figure s8c).anal.calcd for c
92h54
n8:c,86.91;h,4.28;n,8.81.found:c,87.05;h,4.35;n,8.89.
[0045]
(5)双拱门钴卟啉的合成:
[0046]
在甲醇和二氯甲烷的混合溶液中加入双拱门卟啉和醋酸钴盐,双拱门卟啉和醋酸钴盐的物质的量之比为1:8,回流反应24h后,水洗处理然后进行柱层析纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门钴卟啉;hrms:calcd for c
92h52
con8,1327.3641;found,1327.3651(figure s9).anal.calcd for c
92h52
con8:c,83.18;h,3.95;n,8.44.found:c,83.32;h,4.01;n,8.52。
[0047]
实施例2
[0048]
新型金属卟啉化合物的制备方法,包括如下步骤:
[0049]
(1)2-(对溴苯)-1,10-菲罗啉的合成:
[0050]
将1,10-菲罗啉和对二溴苯在乙醚溶液里混合,二者物质的量之比为1:1,加入正丁基锂作为催化剂进行催化,正丁基锂与反应物总物质的量之比为1.2:1,在室温下反应12h,停止反应后,水洗后过柱纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2-(对溴苯)-1,10-菲罗啉;1h nmr(400mhz,cdcl3):9.23(dd,1h),8.32-8.18(m,4h),8.05(d,1h),7.78(d,2h),7.65(dd,2h)(figure s1).hrms:calcd for c18h12n2br[m h] ,335.0178;found,335.0176(figure s2c).anal.calcd.for c18h11n2br:c,64.50;h,3.31;n,8.36.found:c,64.63;h,3.52;n,8.51.
[0051]
(2)2,9-双(对溴苯)-1,10-菲罗啉的合成:
[0052]
将步骤(1)制备的2-(对溴苯)-1,10-菲罗啉和对二溴苯在乙醚溶液中混合,二者物质的量之比为1:5,加入正丁基锂在室温下反应12h,正丁基锂与反应物总物质的量之比为1.2:1,然后进行纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对溴苯)-1,10-菲罗啉;1h nmr(400mhz,cdcl3):8.32(dd,6h),8.13(d,2h),7.81(s,2h),7.72(d,4h)(figure s3).,hrms:calcd for c
24h15
n2br2[m h]
,490.9577;found,490.9569(figure s4c).anal.calcd.for c
24h14
n2br2:c,58.81;h,2.88;n,5.71.found:c,59.05;h,2.93;n,5.79.
[0053]
(3)2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的合成:
[0054]
将步骤(2)制备的2-(对溴苯)-1,10-菲罗啉和2-醛基苯硼酸在甲醇溶液中混合,二者物质的量之比为1:5,加入过量四三苯基膦二氯化钯作为催化剂,四三苯基膦二氯化钯与反应物总物质的量之比为1:1,加入碳酸钠作为碱,碳酸钠与2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的物质的量之比为2:1,在70℃下回流12h后,用含高浓度nh4oh(10ml)的na2co3(2mol/l,100ml)提取两次,反应结束后水洗,然后与二氧化锰共同加入至二氯甲烷中,室温反应30min后,进行过柱处理纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉;1hnmr(400mhz,(cd3)2so):10.04(s,2h),8.68(dd,6h),8.53(d,2h),8.07(s,2h),7.98(d,2h),7.81(t,2h)7.72(d,4h),7.65(dd,4h)(figure s5).hrms:calcd for c
38h24
n2o2na[m na]
,563.1717;found,563.1730(figure s6c).anal.calcd.for c
38h24
n2o2:c,84.42;h,4.47;n,5.18.found:c,84.65;h,4.55;n,5.37.
[0055]
(4)双拱门卟啉的合成:
[0056]
在装有磁性搅拌棒的烧瓶中,加入2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉和吡咯,二者物质的量之比为1:2,然后在n2下加入dcm(400ml),然后加入三氟乙酸,三氟乙酸与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉的物质的量之比为0.01:1.2,在室温下搅拌20h后,加入2,3-二氯-5,6-双氰诺-1,4-苯醌,2,3-二氯-5,6-双氰诺-1,4-苯
醌与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉等物质的量,避光回流2h,然后用三乙胺中和,加水三次萃取后,用mgso4干燥有机相,然后使用柱层析进行纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门卟啉;1h nmr(400mhz,cdcl3):8.76(d,2h),8.69-8.54(m,6h),8.37(td,2h),8.16(d,1h),8.08(s,1h),8.06(dd,2h),7.99(d,2h),7.85-7.78(m,6h),7.75-7.60(m,7h),7.59-7.51(m,7h),7.08(td,4h),6.91(d,4h),6.84(d,2h),6.64(d,6h),-2.92(s,2h)(figure s7).hrms:calcd for c
92h55
n8[m h] ,1271.4544;found,1271.4542(figure s8c).anal.calcd for c
92h54
n8:c,86.91;h,4.28;n,8.81.found:c,87.05;h,4.35;n,8.89.
[0057]
(5)双拱门钴卟啉的合成:
[0058]
在甲醇和二氯甲烷的混合溶液中加入双拱门卟啉和醋酸钴盐,双拱门卟啉和醋酸钴盐的物质的量之比为1:5,回流反应24h后,水洗处理然后进行柱层析纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门钴卟啉;hrms:calcd for c
92h52
con8,1327.3641;found,1327.3651(figure s9).anal.calcd for c
92h52
con8:c,83.18;h,3.95;n,8.44.found:c,83.32;h,4.01;n,8.52。
[0059]
实施例3
[0060]
新型金属卟啉化合物的制备方法,包括如下步骤:
[0061]
(1)2-(对溴苯)-1,10-菲罗啉的合成:
[0062]
将1,10-菲罗啉和对二溴苯在乙醚溶液里混合,二者物质的量之比为1:3,加入正丁基锂作为催化剂进行催化,正丁基锂与反应物总物质的量之比为1.5:1,在室温下反应8h,停止反应后,水洗后过柱纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2-(对溴苯)-1,10-菲罗啉;1h nmr(400mhz,cdcl3):9.23(dd,1h),8.32-8.18(m,4h),8.05(d,1h),7.78(d,2h),7.65(dd,2h)(figure s1).hrms:calcd for c18h12n2br[m h] ,335.0178;found,335.0176(figure s2c).anal.calcd.for c18h11n2br:c,64.50;h,3.31;n,8.36.found:c,64.63;h,3.52;n,8.51.
[0063]
(2)2,9-双(对溴苯)-1,10-菲罗啉的合成:
[0064]
将步骤(1)制备的2-(对溴苯)-1,10-菲罗啉和对二溴苯在乙醚溶液中混合,二者物质的量之比为1:1,加入正丁基锂在室温下反应8h,正丁基锂与反应物总物质的量之比为1.5:1,然后进行纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对溴苯)-1,10-菲罗啉;1h nmr(400mhz,cdcl3):8.32(dd,6h),8.13(d,2h),7.81(s,2h),7.72(d,4h)(figure s3).,hrms:calcd for c
24h15
n2br2[m h]
,490.9577;found,490.9569(figure s4c).anal.calcd.for c
24h14
n2br2:c,58.81;h,2.88;n,5.71.found:c,59.05;h,2.93;n,5.79.
[0065]
(3)2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的合成:
[0066]
将步骤(2)制备的2,9-双(对溴苯)-1,10-菲罗啉和2-醛基苯硼酸在甲醇溶液中混合,二者物质的量之比为1:1,加入过量四三苯基膦二氯化钯作为催化剂,四三苯基膦二氯化钯与反应物总物质的量之比为5:1,加入碳酸钠作为碱,碳酸钠与2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉的物质的量之比为2:1,在80℃下回流8h后,用含高浓度nh4oh(10ml)的na2co3(2mol/l,100ml)提取两次,反应结束后水洗,然后与二氧化锰共同加入至二氯甲烷中,室温反应40min后,进行过柱处理纯化,纯化试剂为:dcm/meoh=95:5,v/v,得2,9-双(对-(2-甲酰苯基)苯基)-l,10-菲罗啉;1hnmr(400mhz,(cd3)2so):10.04(s,2h),8.68
(dd,6h),8.53(d,2h),8.07(s,2h),7.98(d,2h),7.81(t,2h)7.72(d,4h),7.65(dd,4h)(figure s5).hrms:calcd for c
38h24
n2o2na[m na]
,563.1717;found,563.1730(figure s6c).anal.calcd.for c
38h24
n2o2:c,84.42;h,4.47;n,5.18.found:c,84.65;h,4.55;n,5.37.
[0067]
(4)双拱门卟啉的合成:
[0068]
在装有磁性搅拌棒的烧瓶中,加入2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉和吡咯,二者物质的量之比为1:3,然后在n2下加入dcm(400ml),然后加入三氟乙酸,三氟乙酸与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉的物质的量之比为0.01:1.5,在室温下搅拌20h后,加入2,3-二氯-5,6-双氰诺-1,4-苯醌,2,3-二氯-5,6-双氰诺-1,4-苯醌与2,9-双(对-(2-甲酰基苯基)苯基)-l,10-邻菲罗啉等物质的量,避光回流2h,然后用三乙胺中和,加水三次萃取后,用mgso4干燥有机相,然后使用柱层析进行纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门卟啉;1h nmr(400mhz,cdcl3):8.76(d,2h),8.69-8.54(m,6h),8.37(td,2h),8.16(d,1h),8.08(s,1h),8.06(dd,2h),7.99(d,2h),7.85-7.78(m,6h),7.75-7.60(m,7h),7.59-7.51(m,7h),7.08(td,4h),6.91(d,4h),6.84(d,2h),6.64(d,6h),-2.92(s,2h)(figure s7).hrms:calcd for c
92h55
n8[m h] ,1271.4544;found,1271.4542(figure s8c).anal.calcd for c
92h54
n8:c,86.91;h,4.28;n,8.81.found:c,87.05;h,4.35;n,8.89.
[0069]
(5)双拱门钴卟啉的合成:
[0070]
在甲醇和二氯甲烷的混合溶液中加入双拱门卟啉和醋酸钴盐,双拱门卟啉和醋酸钴盐的物质的量之比为1:10,回流反应24h后,水洗处理然后进行柱层析纯化,纯化试剂为:dcm/meoh=200:1,v/v,得双拱门钴卟啉;hrms:calcd for c
92h52
con8,1327.3641;found,1327.3651(figure s9).anal.calcd for c
92h52
con8:c,83.18;h,3.95;n,8.44.found:c,83.32;h,4.01;n,8.52。
[0071]
对比例1
[0072]
现有技术的无位阻的苯基卟啉,其结构式为:其制备方法包括如下步骤:
[0073]
(1)将吡咯(2.12g,20mmol)和苯甲醛(1.34g,20mmol)在丙酸(75ml)中回流反应30min,制得卟啉配体;
[0074]
(2)将步骤(1)的卟啉配体再与醋酸钴在dcm/meoh混合溶液中回流反应3h,得到配合物钴卟啉。
[0075]
本技术制得的新型金属卟啉化合物均为位阻型卟啉,下面以实施例1制得的新型金属卟啉化合物为例,与对比例1的无位阻的苯基卟啉进行orr性能的对比,通过旋转圆盘电极进行测试,具体为:将催化剂与碳黑等质量混合后,分散在1ml乙醇中得到浆料,然后将该浆料滴涂于电极表面并晾干,再进行氧气还原测试;
[0076]
我们发现在相同的测试条件下,使用rrde电极测试,对比例1的无位阻的苯基卟啉(传统的苯基卟啉)是一个复杂的三电子过程,产物通常包含水和双氧水,而本技术实施例1制得的新型金属卟啉化合物可以成功的进行两电子过程,得到产物是双氧水,这达到了我们设计的目的。
[0077]
图2结果表明,通过旋转圆盘电极(rrde)测试发现,其在0.1m koh中电催化氧气还原的电子数大约是3,说明普通钴卟啉存在氧气还原的双分子机制(即四电子过程)。
[0078]
图3结果表明,通过旋转圆盘电极(rrde)测试发现,其在0.1m koh中电催化氧气还原的电子数大约是2,说明本技术设计合成的钴卟啉能够通过位阻效应阻止双分子机制的氧气还原发生(即四电子过程)。
[0079]
图4为本发明实施例1制得的双拱门卟啉配体的氢谱图,结果表明本技术制得了所需结构的双拱门卟啉配体。
[0080]
本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。