1.本发明涉及对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯的制备方法,属于有机合成领域。
背景技术:
2.聚乙二醇是一类由乙二醇聚合而成的化合物,简称peg-n,其中n代表聚合度。聚乙二醇及其相关衍生物如今在医药领域尤其是药物输送领域的应用已成为研究的热点。由于聚乙二醇具有界面张力低、生物分子相容性好、无毒、安全无刺激性等特点,并且具有很好的润滑性,保湿性,因此选择将聚乙二醇与其他类型的高分子聚合物用共混或共聚的方式固定并附着在一些高分子聚合物材料表面结构上,来实现聚乙二醇作为药物载体,实现药物输送的功能,此外,通过合理地利用聚乙二醇的性质,可以有效的提高药物的药效,比如:通过使所修饰的药物分子质量增大,实现药物的肾清除率降低,半衰期延长;利用聚乙二醇本身具有的优良的水溶性,可对药物在水溶液中的溶解度起到一定的提高的作用;聚乙二醇分散在药物表面可以起到屏蔽和位阻效应,降低酶解作用,提高了药物分子的稳定性。
3.由于分子尺寸与分子量的区别,不同聚合度的聚乙二醇在物理性质上表现出显著的差异,进而影响其的应用。高纯度的低聚合度的聚乙二醇具有显著的单分散性,具体表现在可结晶,可通过分析方法得到确定的晶体结构,此外,低聚合度的聚乙二醇大都具有强水溶性,亦可溶于如二氯甲烷、三氯甲烷、丙酮等有机溶剂,但在乙醚等小极性溶剂中相对溶解度较差。随着聚合度增大,聚乙二醇的水溶性、脂溶性、密度、都发生明显的变化,此外,高分子量的聚乙二醇闪点与粘度也会大大增加,从而无法结晶。值得注意的是除了明显变化的物理性质,高分子量的聚乙二醇因其尺寸大和反应位点少等特点,从而表现出显著的化学惰性。
4.基于聚乙二醇的广泛用途和本身固有的优点,设计并合成具有精确聚合度的双官能团化的聚乙二醇具有重要的意义。
技术实现要素:
5.为了解决上述问题,本发明提供了对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯的制备方法。主要分为低聚乙二醇的保护、聚合度增加、迈克尔反应/烷基化反应、脱三苯甲基保护基、酯化等五步,具体如下:碱性条件下低聚乙二醇和三苯氯甲烷作用得到三苯甲基保护的低聚乙二醇;接着在强碱条件下三苯甲基保护低聚乙二醇与四聚乙二醇磺酸内酯反应发生增链反应后经过酸化处理得到聚合度增大的三苯甲基保护低聚乙二醇;接着在非质子性溶剂中,碱性条件下与丙烯酸叔丁酯发生分子间迈克尔加成反应,或与溴乙酸叔丁酯反应生成三苯甲基保护低聚乙二醇丙酸叔丁酯;随后催化量酸作用下三苯甲基保护低聚乙二醇丙酸叔丁酯脱除三苯甲基保护基生成单官能团化的低聚乙二醇;最后单官能团化的低聚乙二醇与对甲基苯磺酰氯反应即可得到甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯。该合成方法操作简单,合成成本低廉,反应条件温和,易于大规模生产,收率高,并且在合成步骤中
不涉及到金属催化剂的使用。
6.本发明中提供对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯的制备方法,反应方程式表示为:
[0007][0008]
其中:n=1,2,3,4;m=0,1;
[0009]
包括以下步骤:
[0010]
第一步、碱性条件下,低聚乙二醇2和三苯基氯甲烷,在催化量4-二甲氨基吡啶存在下,反应得到三苯甲基保护低聚乙二醇3;
[0011]
第二步、用氢化钠处理后,三苯甲基保护低聚乙二醇与四聚乙二醇磺酸内酯4反应得到链增长的磺酸盐,经硫酸酸化后得到三苯甲基保护聚乙二醇5;
[0012]
第三步、在氢化钠作用下,三苯甲基保护聚乙二醇5与丙烯酸叔丁酯6a发生迈克尔加成反应,或与溴乙酸叔丁酯6b发生氧烷基化反应生成三苯甲基保护聚乙二醇丙酸叔丁酯7;
[0013]
第四步、在催化量对甲基苯磺酸存在下,三苯甲基保护聚乙二醇丙酸叔丁酯7脱除三苯甲基保护基,生成一端为游离羟基聚乙二醇丙酸叔丁酯8;
[0014]
第五步、在三乙胺和4-二甲氨基吡啶存在下,一端为游离羟基聚乙二醇丙酸叔丁酯8与对甲基苯磺酰氯发生磺酰酯化反应,生成对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯1。
[0015]
进一步地,在上述技术方案中,第一步中,低聚乙二醇与4-二甲氨基吡啶摩尔比为1:0.1-0.3。
[0016]
进一步地,在上述技术方案中,第一步中,低聚乙二醇与三苯基氯甲烷摩尔比为1:1.05-1.15。
[0017]
进一步地,在上述技术方案中,第一步中,低聚三苯基氯甲烷/二氯甲烷溶液缓慢滴加到低聚乙二醇/二氯甲烷溶液中,滴加速度为3-5ml/min。
[0018]
进一步地,在上述技术方案中,第二步中,三苯甲基保护低聚乙二醇3与四聚乙二醇磺酸内酯4摩尔比为1:1.2-2.0。
[0019]
进一步地,在上述技术方案中,第二步中,三苯甲基保护低聚乙二醇3与氢化钠摩尔比为1:1.5-2.5。
[0020]
进一步地,在上述技术方案中,第二步中,三苯甲基保护低聚乙二醇3与浓硫酸摩尔比为1:1.0-1.2,反应时间为5-20min。
[0021]
进一步地,在上述技术方案中,第三步中,对于6a反应物而言,三苯甲基保护聚乙二醇5与氢化钠摩尔比为1:0.1-0.5。对于6b反应物而言,三苯甲基保护聚乙二醇5与氢化钠摩尔比为1:1.2-2.0。
[0022]
进一步地,在上述技术方案中,第三步中,三苯甲基保护聚乙二醇5与丙烯酸叔丁酯6a摩尔比为1:1.2-1.8;三苯甲基保护聚乙二醇5与溴乙酸叔丁酯6b摩尔比为1:1.2-1.8。
[0023]
进一步地,在上述技术方案中,第四步中,三苯甲基保护聚乙二醇丙酸叔丁酯7与对甲基苯磺酸摩尔比为1:0.1-0.5。
[0024]
进一步地,在上述技术方案中,第四步中,脱三苯甲基保护基反应时间为1-3h,反应温度为25-30℃。
[0025]
进一步地,在上述技术方案中,第五步中,一端为游离羟基聚乙二醇丙酸叔丁酯8与4-二甲氨基吡啶摩尔比为1:0.1-0.3。
[0026]
进一步地,在上述技术方案中,第五步中,一端为游离羟基聚乙二醇丙酸叔丁酯8与三乙胺摩尔比为1:1.3-2.0。
[0027]
进一步地,在上述技术方案中,第五步中,一端为游离羟基聚乙二醇丙酸叔丁酯8与对甲基苯磺酰氯摩尔比为1:1.1-1.3。
[0028]
进一步地,在上述技术方案中,第五步中,反应温度为0-30℃,反应时间为4-6小时。
[0029]
进一步地,在上述技术方案中,其特征在于:第五步后处理为,反应结束后,反应体系用饱和氯化铵淬灭,并用二氯甲烷萃取三次,有机相用饱和实验室洗涤后用无水硫酸钠干燥,过滤,旋干,柱层析纯化,得到对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯。
[0030]
发明有益效果:
[0031]
本发明提供的制备对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯的方法简单,便于操作,所使用的原料便宜易得,且无需金属催化剂的使用,合成成本低廉,反应条件温和,易于大规模生产;得到的目标产物的产率高,纯度高;制备过程也较为环保,反应溶剂可以回收并重复利用。
附图说明
[0032]
图1、图2为本发明实施例1中第一步合成的单三苯甲基保护四聚乙二醇3a的核磁共振氢谱、碳谱图;
[0033]
图3、图4为本发明实施例1中第二步合成的单三苯甲基保护八聚乙二醇5a的核磁共振氢谱、碳谱图;
[0034]
图5、图6为本发明实施例1中第三步合成的三苯甲基保护八聚乙二醇丙酸叔丁酯7a的核磁共振氢谱、碳谱图。
[0035]
图7、图8为本发明实施例1中第四步合成的一端为游离羟基八聚乙二醇丙酸叔丁酯8a的核磁共振氢谱、碳谱图。
[0036]
图9、图10为本发明实施例1中第五步合成的对甲苯磺酰氧基取代八聚乙二醇丙酸叔丁酯1a的核磁共振氢谱、碳谱图。
具体实施方式
[0037]
以下通过具体实例进一步描述本发明。不过这些实例仅仅是范例性的,并不局限于本发明的保护范围仅仅为实施例。
[0038]
在下述实施例中,所使用到的试剂、材料以及仪器如没有特殊的说明,均为常规试剂、常规材料以及常规仪器,均可商购获得,其中所涉及的试剂也可通过常规合成方法合成获得。
[0039]
实施例1
[0040]
第一步、单三苯甲基保护四聚乙二醇3的制备
[0041]
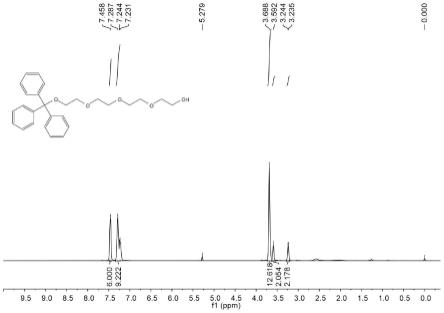
依次向反应瓶中加入25.9ml(150mmol,3.0eq)四聚乙二醇,250ml二氯甲烷,14ml(100mmol,2.0eq)三乙胺及610.9mg(5mmol,0.1eq)dmap于反应瓶中,然后用水泵抽换氮气三次,在40℃,氮气条件下逐滴加入三苯基氯甲烷(13.9338g,50mmol,1.0eq)二氯甲烷(50ml)溶液(滴加2h)。滴加完毕升温至55℃回流。12h后,tlc先用pe/ea=10/1监测原料是否反应完全,再用pe/ea=1/1监测产物分布。反应完毕后用饱和食盐水淬灭,减压旋除大约250ml dcm,剩余残留物用dcm萃取三次,合并有机相饱和食盐水洗,硫酸钠干燥,过滤旋干,柱层析得产物3a(20.4468g,94%)。1h nmr(400mhz,cdcl3)δ7.458(s,6h),7.287(s,6h),7.231(s,3h),3.688(s,12h),3.592(s,2h),3.240(d,j=3.6hz,2h).
13
c nmr(100mhz,cdcl3)δ144.12,128.74,127.82,126.98,86.57,72.53,70.81,70.76,70.73,70.43,63.34,61.79.
[0042]
第二步、单三苯甲基保护八聚乙二醇5a的制备
[0043]
氮气氛围,0℃条件下,将12mlthf和312mg(7.8mmol,1.5eq)氢化钠依次加入反应瓶,然后加入3a(2.2684g,5.2mmol,1.0eq)/thf(8ml)溶液。保持0℃15分钟后逐滴滴入四聚乙二醇磺酸内酯4(1.9986g,7.8mmol,1.5eq)/thf(5ml)溶液。滴加完毕后,0℃持续15min后升至室温。5h后tlc监测原料反应完毕,氮气条件下依次慢慢加入0.2ml(10.2mmol,2.0eq)水和0.28ml(5.2mmol,1.0eq)硫酸(98%)。搅拌5min tlc监测反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,dcm萃取三次,无水硫酸钠干燥,过滤旋干,柱层析得产物5a(2.499g,79%)。1h nmr(cdcl3,400mhz):δ7.466(s,6h),7.288-7.236(m,9h),3.713-3.608(m,32h),3.232(br,1h).
13
c nmr(100mhz,cdcl3)δ144.12,128.72,127.79,126.94,86.52,72.69,72.64,71.91,70.78,70.70,70.67,70.61,70.55,70.27,69.81,66.71,63.31,61.71,59.06,15.13.
[0044]
第三步、三苯甲基保护八聚乙二醇丙酸叔丁酯7a的制备
[0045]
在0℃,氮气条件下,依次向反应瓶中加入2.499g化合物5a(4.1mmol,1.0eq)、7.6ml thf和16.4mg(0.41mmol,0.1eq)氢化钠。10min后升至室温并搅拌2h,随后反应体系降温至0℃,并向体系中逐滴滴加0.77ml(5.33mmol,1.3eq)丙烯酸叔丁酯,0℃持续10分钟后升至室温并搅拌过夜。反应完毕后用水淬灭,乙酸乙酯萃取三次,饱和食盐水洗,无水硫酸钠干燥,过滤旋干,柱层析得产物7a(2.1523g,71%)。1h nmr(cdcl3,400mhz):δ7.463-7.454(d,j=3.6hz,6h),7.287-7.235(m,9h),3.677-3.634(m,32h),3.232(s,2h),2.507(s,2h),1.445(s,9h).
13
c nmr(100mhz,cdcl3)δ170.98,144.13,128.73,127.80,126.95,86.53,80.57,70.80,70.72,70.68,70.57,70.51,70.38,66.91,63.32,53.49,36.25,28.12.
[0046]
第四步、一端为游离羟基的八聚乙二醇丙酸叔丁酯8a的制备
[0047]
在氮气条件下,将903.3mg化合物7a(1.22mmol,1.0eq)溶解到meoh/thf=1/1(3ml/3ml)混合溶液,然后再向混合液中加入23.2mg(0.122mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。tlc检测反应完毕,用碳酸氢钠淬灭后再在减压条件下旋去溶剂,残留物用dcm萃取三次,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物8a(462.6mg,76%)。1h nmr(cdcl3,400mhz):δ3.720-3.621(m,34h),2.826(br,1h),2.516-2.502(d,j=5.6hz,2h),1.448(s,9h).
13
c nmr(100mhz,cdcl3)δ170.96,80.54,72.66,70.55,70.36,70.25,66.88,61.68,36.23,28.10.
[0048]
第五步、对甲苯磺酰氧基取代八聚乙二醇丙酸叔丁酯1a的制备
[0049]
氮气条件下,依次向圆底烧瓶加入1.1224g化合物8a(2.3mmol,1.0eq)、421.5mg(3.45mmol,1.5eq)dmap和3ml dcm,然后在0℃条件下逐滴加入对甲苯磺酰氯(526.3mg,2.76mmol,1.2eq)dcm(3ml)溶液,滴加完毕后保持该温度反应10min后升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,dcm萃取三次,有机相饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物1a(1.3158g,88%)。1h nmr(cdcl3,400mhz):δ7.805-7.795(d,j=4hz,2h),7.355(s,2h),4.160(s,2h),3.701-3.586(m,34h),2.516-2.503(d,j=5.2hz,2h),2.453(s,3h),1.447(s,9h).
13
c nmr(100mhz,cdcl3)δ170.97,144.85,132.92,129.86,128.02,80.57,70.76,70.57,70.52,70.38,69.28,68.69,66.91,53.50,36.25,29.74,29.36,28.12,21.70.
[0050]
实施例2
[0051]
第一步、单三苯甲基保护三聚乙二醇的制备
[0052]
依次向反应瓶中加入20.5ml(150mmol,3.0eq)三聚乙二醇,250ml二氯甲烷、14ml(100mmol,2.0eq)三乙胺及610.9mg(5mmol,0.1eq)dmap于反应瓶中,然后用水泵抽换氮气三次,在40℃氮气条件下逐滴加入13.9338g(50mmol,1.0eq)三苯基氯甲烷/二氯甲烷(50ml)溶液(滴加2h)。滴加完毕升温至55℃回流。12h后,tlc监测反应完毕,饱和食盐水淬灭,减压旋除大约250ml dcm,剩余残留物用dcm萃取三次,合并有机相饱和食盐水洗涤,硫酸钠干燥,过滤旋干,柱层析得19.5262g产物3b,产率99%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ7.458-7.473(m,6h),7.229-7.294(m,9h),3.699-3.724(m,8h),3.633(d,j=2.4hz,2h),3.255(d,j=4.0hz,2h).
13
c nmr(100mhz,cdcl3)δ144.08,128.73,127.82,127.00,86.61,72.53,70.86,70.73,70.59,63.29,61.87.
[0053]
第二步、单三苯甲基保护七聚乙二醇的制备
[0054]
氮气氛围,0℃条件下,将15ml四氢呋喃和312mg(7.8mmol,1.5eq)氢化钠依次加入反应瓶,然后加入2.0394g(5.2mmol,1.0eq)单三苯甲基保护三聚乙二醇3b/四氢呋喃(10ml)溶液。保持0℃15分钟后逐滴滴入1.9986g(7.8mmol,1.5eq)四聚乙二醇磺酸内酯4/四氢呋喃(5ml)溶液。滴加完毕后,0℃持续15min后升至室温。5htlc监测原料反应完毕,氮气条件下依次慢慢加入0.2ml(10.2mmol,2.0eq)水和0.28ml(5.2mmol,1.0eq)硫酸(98%)。搅拌5min tlc监测反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,二氯甲烷萃取三次,有机相用无水硫酸钠干燥,过滤,旋干,柱层析得2.2g产物5b,产率74.5%,高效液相色谱(hplc)检测产品纯度98%。
[0055]
第三步、三苯甲基保护七聚乙二醇丙酸叔丁酯的制备
[0056]
在0℃氮气条件下,依次向反应瓶中加入496.6mg(0.8738mmol,1.0eq)单三苯甲基保护七聚乙二醇5b、2ml四氢呋喃和3.5mg(0.0874mmol,0.1eq)氢化钠。10min后升至室温并搅拌2h,随后反应体系降温至0℃,并向体系中逐滴滴加0.165ml(1.1359mmol,1.3eq)丙烯酸叔丁酯6a,0℃持续10分钟后升至室温并搅拌过夜。反应完毕后用水淬灭,乙酸乙酯萃取三次,有机相用饱和食盐水洗,合并有机相用无水硫酸钠干燥,过滤,旋干,柱层析得572.0mg产物7aa,产氯94%,高效液相色谱(hplc)检测产品纯度98%。
[0057]
第四步、一端为游离羟基的七聚乙二醇丙酸叔丁酯的制备
[0058]
氮气条件下,将1.5316g(2.2mmol,1.0eq)三苯甲基保护聚乙二醇丙酸叔丁酯7aa溶解到甲醇/四氢呋喃=1/1(5.4ml/5.4ml)混合溶液,然后再向混合液中加入41.8mg(0.22mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。tlc检测反应完毕,碳酸氢钠淬灭后再在减压条件下旋去溶剂,残留物dcm萃取三次,合并有机相并无水硫酸钠干燥,过滤,旋干,柱层析得751.6mg产物8aa,产率75%,高效液相色谱(hplc)检测产品纯度98%。
[0059]
第五步、对甲苯磺酰氧基取代七聚乙二醇丙酸叔丁酯的制备
[0060]
氮气条件下,依次向圆底烧瓶加入280.5mg(0.62mmol,1.0eq)一端为游离羟基聚乙二醇丙酸叔丁酯8aa,113.2mg(0.93mmol,1.5eq)4-二甲氨基吡啶和3.3ml二氯甲烷,然后再0℃条件下逐滴加入141.3mg(0.74mmol,1.2eq)对甲苯磺酰氯/二氯甲烷(1.1ml)溶液,滴加完毕后保持该温度反应10min,升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,二氯甲烷萃取三次,有机相饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物,产率88%,高效液相色谱(hplc)检测产品纯度98%。
[0061]
实施例3
[0062]
第一步、单三苯甲基保护二聚乙二醇的制备
[0063]
依次向反应瓶中加入5.7ml(60mmol,3.0eq)二聚乙二醇,100ml二氯甲烷、5.5ml(40mmol,2.0eq)三乙胺及244.34mg(2mmol,0.1eq)dmap于反应瓶中,然后用水泵抽换氮气三次,在40℃,氮气条件下逐滴加入5.5755g(50mmol,1.0eq)三苯基氯甲烷/二氯甲烷(20ml)溶液(滴加1h)。滴加完毕升温至55℃回流。12h后tlc先用pe/ea=10/1监测原料是否反应完全,再用pe/ea=3/1监测产物分布。反应完毕后用饱和食盐水淬灭,减压旋除大约100ml dcm,剩余残留物用dcm萃取三次,合并有机相并用饱和食盐水洗涤,硫酸钠干燥,过滤,旋干,柱层析得产物单三苯甲基保护聚乙二醇6.4133g,产率92.1%,高效液相色谱(hplc)检测产品纯度为98%。
[0064]
第二步、单三苯甲基保护六聚乙二醇的制备
[0065]
氮气氛围,0℃条件下,将7.4mlthf和156mg(3.9mmol,1.5eq)氢化钠依次加入反应瓶,然后加入905.2mg(5.2mmol,1.0eq)单三苯甲基保护2聚乙二醇的thf(5ml)溶液。保持0℃下15分钟后逐滴滴入999.3mg(7.8mmol,1.5eq)四聚乙二醇磺酸内酯/thf(3ml)溶液。滴加完毕后,0℃持续15min后升至室温。5h后tlc监测。原料反应完毕后,氮气条件下依次慢慢加入0.1ml(5.2mmol,2.0eq)水和0.14ml(2.6mmol,1.0eq)硫酸(98%)。搅拌5min后用tlc监测。反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,dcm萃取三次,有机相无水硫酸钠干燥,过滤旋干,柱层析得产物1.0095g,经计算产率为74%,经高效液相色谱(hplc)检测产品纯度为98%。第三步、三苯甲基保护六聚乙二醇丙酸叔丁酯的制备
[0066]
在0℃,氮气条件下,依次向反应瓶中加入1.0095g(1.9mmol,1.0eq)单三苯甲基保
护六聚乙二醇、4mlthf和7.6mg(0.19mmol,0.1eq)氢化钠。10min后升至室温并搅拌2h,随后反应体系降温至0℃,并向体系中逐滴滴加0.33ml(2.28mmol,1.2eq)丙烯酸叔丁酯,0℃持续10分钟后升至室温并搅拌过夜。反应完毕后用水淬灭,乙酸乙酯萃取三次,有机相用饱和食盐水洗,合并有机相用无水硫酸钠干燥,过滤,旋干,柱层析得产物793.6mg,产率64%,高效液相色谱(hplc)检测产品纯度98%。
[0067]
第四步、一端为游离羟基的六聚乙二醇丙酸叔丁酯的制备
[0068]
氮气条件下,将793.6mg(1.22mmol,1.0eq)三苯甲基保护聚乙二醇丙酸叔丁酯溶解到meoh/thf=1/1(3ml/3ml)混合溶液,然后再向混合液中加入23.2mg(0.122mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。tlc检测反应完毕,碳酸氢钠淬灭再在减压条件下蒸去溶剂,残留物dcm萃取三次,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物436.5mg,产率87%,经高效液相色谱(h plc)检测产品纯度98%。
[0069]
第五步、对甲苯磺酰氧基取代低聚乙二醇丙酸叔丁酯的制备
[0070]
氮气条件下,依次向圆底烧瓶加入508.7mg(1.24mmol,1.0eq)一端为游离羟基六聚乙二醇丙酸叔丁酯,227.2mg(1.86mmol,1.5eq)dmap和3ml dcm,然后再0℃条件下逐滴加入283.8mg(2.76mmol,1.2eq)对甲苯磺酰氯/dcm(0.8ml)溶液,滴加完毕后保持该温度反应10min后升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,dcm萃取三次,有机相饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物552.7mg,产率79%,高效液相色谱(hplc)检测产品纯度98%。
[0071]
实施例4
[0072]
第一步、单三苯甲基保护的四聚乙二醇的制备
[0073]
依次向反应瓶中加入25.9ml(150mmol,3.0eq)四聚乙二醇,250ml二氯甲烷、14ml(100mmol,2.0eq)三乙胺及610.9mg(5mmol,0.1eq)4-二甲氨基吡啶于反应瓶中,然后用水泵抽换氮气三次,在40℃,氮气条件下逐滴加入三苯基氯甲烷(13.9338g,50mmol,1.0eq)/二氯甲烷(50ml)溶液(滴加2h)。滴加完毕升温至55℃回流。12h后tlc监测反应完毕,饱和食盐水淬灭,减压旋除大约250ml二氯甲烷,剩余残留物用二氯甲烷萃取三次,合并有机相饱和食盐水洗涤,硫酸钠干燥,过滤旋干,柱层析得产物四聚乙二醇20.4468g,产率94%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ7.458(s,6h),7.287(s,6h),7.231(s,3h),3.688(s,12h),3.592(s,2h),3.240(d,j=3.6hz,2h).
13
c nmr(100mhz,cdcl3)δ144.12,128.74,127.82,126.98,86.57,72.53,70.81,70.76,70.73,70.43,63.34,61.79.
[0074]
第二步、单三苯甲基保护的八聚乙二醇的制备
[0075]
氮气氛围,0℃条件下,将12ml四氢呋喃和312mg(7.8mmol,1.5eq)氢化钠依次加入反应瓶,然后加入单三苯甲基保护四聚乙二醇(2.2684g,5.2mmol,1.0eq)/四氢呋喃(8ml)溶液。保持0℃15分钟后逐滴滴入四聚乙二醇磺酸内酯(1.9986g,7.8mmol,1.5eq)/四氢呋喃(5ml)溶液。滴加完毕后,0℃持续15min后升至室温。5h后tlc监测原料反应完毕,氮气条件下依次慢慢加入0.2ml(10.2mmol,2.0eq)水和0.28ml(5.2mmol,1.0eq)硫酸(98%)。搅拌5min tlc监测,反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,二氯甲烷萃取三次,有机相用无水硫酸钠干燥,过滤,旋干,柱层析得产物2.499g,产率79%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ7.466(s,6h),7.288-7.236(m,9h),
3.713-3.608(m,32h),3.232(br,1h).
13
c nmr(100mhz,cdcl3)δ144.12,128.72,127.79,126.94,86.52,72.69,72.64,71.91,70.78,70.70,70.67,70.61,70.55,70.27,69.81,66.71,63.31,61.71,59.06,15.13.
[0076]
第三步、三苯甲基保护的八聚乙二醇乙酸叔丁酯的制备
[0077]
在0℃氮气条件下,依次向反应瓶中加入866.2mg(1.4mmol,1.0eq)单三苯甲基保护聚乙二醇,2.8ml n,n-二甲基甲酰胺和67.2mg(1.68mmol,1.2eq)氢化钠。10min后向体系中逐滴滴加0.24ml(1.47mmol,1.05eq)/2-溴乙酸叔丁酯,0℃持续10分钟后升至室温并搅拌过夜。反应完毕后用水淬灭,乙酸乙酯萃取三次,有机相用饱和食盐水洗,合并有机相用无水硫酸钠干燥,过滤旋干,柱层析得产物758.2mg,产率75%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ7.465-7.458(d,j=2.8hz,6h),7.287-7.238(m,9h),4.024(s,2h),3.678-3.637(m,30h),3.232(s,2h),1.475(s,9h).
13
c nmr(100mhz,cdcl3)δ144.13,128.73,127.80,126.95,86.52,81.59,70.80,70.73,70.69,70.62,70.58,69.04,63.32,28.14.
[0078]
第四步、一端为游离羟基的八聚乙二醇乙酸叔丁酯的制备
[0079]
氮气条件下,将758.2mg(1.04mmol,1.0eq)三苯甲基保护聚乙二醇丙酸叔丁酯溶解到甲醇/四氢呋喃=1/1(2.5ml/2.5ml)混合溶液,然后再向混合液中加入19.8mg(0.104mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。tlc检测反应完毕,碳酸氢钠淬灭后再在减压条件下旋去溶剂,残留物用二氯甲烷萃取三次,合并有机相并用无水硫酸钠干燥,过滤旋干,柱层析得产物397.8mg,产率79%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ4.066(s,2h),3.707-3.613(m,32h),3.385(s,1h),1.480(s,9h).
13
c nmr(100mhz,cdcl3)δ169.73,81.91,81.59,72.62,70.73,70.58,70.31,69.85,69.04,68.48,66.68,61.75,60.98,53.49,28.13,28.04,15.20.
[0080]
第五步、对甲苯磺酰氧基-八聚乙二醇-乙酸叔丁酯的制备
[0081]
氮气条件下,依次向圆底烧瓶加入397.8mg(0.82mmol,1.0eq)一端为游离羟基八聚乙二醇丙酸叔丁酯,20.4mg(0.164mmol,0.2eq)4-二甲氨基吡啶、3ml二氯甲烷和0.17ml(1.23mmol,1.5eq),然后在0℃条件下逐滴加入187.7mg,(0.984mmol,1.2eq)对甲苯磺酰氯/二氯甲烷(0.5ml)溶液,滴加完毕后保持该温度反应10min后升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,dcm萃取三次,有机相饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物349.1mg,产率67%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ7.804(s,2h),7.352-7.267(d,j=34hz,2h),4.025(s,2h),3.698-3.588(m,32h),2.453(s,3h),1.477(s,9h).
13
c nmr(100mhz,cdcl3)δ169.73,144.85,132.93,129.87,128.03,81.60,70.76,70.73,70.61,70.58,69.29,69.03,68.70,29.74,29.36,28.14,21.71.
[0082]
实施例5
[0083]
第一步、单三苯甲基保护的三聚乙二醇的制备
[0084]
依次向反应瓶中加入20.5ml(150mmol,3.0eq)三聚乙二醇,250ml二氯甲烷、14ml(100mmol,2.0eq)三乙胺及610.9mg(5mmol,0.1eq)dmap于反应瓶中,然后用水泵抽换氮气三次,在40℃氮气条件下逐滴加入13.9338g(50mmol,1.0eq)三苯基氯甲烷/二氯甲烷(50ml)溶液(滴加2h)。滴加完毕升温至55℃回流。12h后tlc监测,待反应完毕后饱和食盐水
淬灭,减压旋除大约250ml dcm,剩余残留物dcm萃取三次,合并有机相饱和食盐水洗涤,硫酸钠干燥,过滤旋干,柱层析得产物三聚乙二醇19.5262g,产率99.6%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ7.458-7.473(m,6h),7.229-7.294(m,9h),3.699-3.724(m,8h),3.633(d,j=2.4hz,2h),3.255(d,j=4.0hz,2h).
13
c nmr(100mhz,cdcl3)δ144.08,128.73,127.82,127.00,86.61,72.53,70.86,70.73,70.59,63.29,61.87.
[0085]
第二步、单三苯甲基保护的七聚乙二醇的制备
[0086]
氮气氛围,0℃条件下,将15ml thf和312mg(7.8mmol,1.5eq)氢化钠依次加入反应瓶,然后加入2.0394g(5.2mmol,1.0eq)单三苯甲基保护聚乙二醇/thf(10ml)溶液。保持0℃下15分钟后逐滴滴入1.9986g(7.8mmol,1.5eq)四聚乙二醇磺酸内酯/thf(5ml)溶液。滴加完毕后,0℃持续15min后升至室温。5h后tlc监测原料反应完毕,氮气条件下依次慢慢加入0.2ml(10.2mmol,2.0eq)水和0.28ml(5.2mmol,1.0eq)硫酸(98%)。搅拌5min后tlc监测反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,dcm萃取三次,有机相无水硫酸钠干燥,过滤旋干,柱层析得产物2.2g,产率74.5%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ7.462(s,6h),7.285-7.231(m,9h),3.677-3.644(m,28h),3.229(br,1h).
13
c nmr(100mhz,cdcl3)δ169.72,144.85,132.91,129.87,128.02,81.59,70.75,70.71,70.61,70.57,69.29,69.02,68.69,28.13,21.71.
[0087]
第三步、三苯甲基保护的七聚乙二醇乙酸叔丁酯的制备
[0088]
在0℃氮气条件下,依次向反应瓶中加入1.1g(1.936mmol,1.0eq)单三苯甲基保护聚乙二醇,3.9ml dmf和92.9mg(2.323mmol,1.2eq)氢化钠。10min后向体系中逐滴滴加0.33ml(2.032mmol,1.05eq)2-溴乙酸叔丁酯,0℃持续10分钟后升至室温并搅拌过夜。反应完毕后用水淬灭,乙酸乙酯萃取三次,有机相用饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物1.0g,产率75.7%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):7.464(s,6h),7.288-7.237(m,9h),4.022(s,1h),3.760(s,1h),3.680-3.643(m,26h),3.236-3.228(d,j=1.6hz,2h),1.480(s,9h).
13
c nmr(100mhz,cdcl3)δ170.98,144.13,128.73,127.80,126.95,86.53,80.57,70.80,70.73,70.69,70.58,70.51,70.38,66.91,63.32,36.25,29.75,28.12.
[0089]
第四步、一端为游离羟基的七聚乙二醇乙酸叔丁酯的制备
[0090]
氮气条件下,将1.0g(1.465mmol,1.0eq)三苯甲基保护聚乙二醇乙酸叔丁酯溶解到meoh/thf=1/1(3.7ml/3.7ml)混合溶液,然后再向混合液中加入27.9mg(0.1465mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。tlc检测反应完毕,碳酸氢钠淬灭后再在减压条件下旋去溶剂,残留物dcm萃取三次,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物885.4mg,产率60.4%,高效液相色谱(hplc)检测产品纯度98%.1h nmr(cdcl3,400mhz):δ4.027(s,2h),3.748-3.610(m,28h),1.477(s,9h).
13
c nmr(100mhz,cdcl3)δ169.67,81.51,72.65,70.85,70.64,70.49,70.18,68.95,68.55,61.57,51.79,28.07.
[0091]
第五步、对甲苯磺酰氧基-七聚乙二醇-乙酸叔丁酯的制备
[0092]
氮气条件下,依次向圆底烧瓶加入389.8mg(0.3898mmol,1.0eq)一端为游离羟基聚乙二醇乙酸叔丁酯、21.6mg(0.1771mmol,0.2eq)dmap、2.2ml dcm和0.2ml(1.3281mmol,1.5eq)三乙胺,然后在0℃条件下逐滴加入202.5mg(1.062mmol,1.2eq)对甲苯磺酰氯/dcm
(0.82ml)溶液,滴加完毕后保持该温度反应10min后升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,dcm萃取三次,有机相饱和食盐水洗,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物331.5mg,产率63.0%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(cdcl3,400mhz):δ7.802(s,2h),7.350(s,2h),4.024(s,2h),3.744-3.586(m,28h),1.476(s,9h).
13
c nmr(100mhz,cdcl3)δ169.72,144.85,132.91,129.87,128.02,81.59,70.75,70.71,70.61,70.57,69.29,69.02,68.69,28.13,21.71.
[0093]
实施例6
[0094]
第一步、单三苯甲基保护的二聚乙二醇的制备
[0095]
依次向反应瓶中加入5.7ml(60mmol,3.0eq)二聚乙二醇,100ml二氯甲烷、5.5ml(40mmol,2.0eq)三乙胺及244.34mg(2mmol,0.1eq)4-二甲氨基吡啶于反应瓶中,然后用水泵抽换氮气三次,在40℃,氮气条件下逐滴加入5.5755g(50mmol,1.0eq)三苯基氯甲烷/二氯甲烷(20ml)溶液(滴加1h)。滴加完毕升温至55℃回流。12h后,tlc监测反应完毕饱和食盐水淬灭,减压旋除大约100ml二氯甲烷,剩余残留物用二氯甲烷萃取三次,合并有机相并用饱和食盐水洗涤,硫酸钠干燥,过滤,旋干,柱层析得产物单三苯甲基保护聚乙二醇6.4133g,经计算产率为92.1%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3),δ7.466(s,6h),7.258-7.298(m,9h),3.632-3.753(m,6h),3.263(s,2h),2.134(br,1h).
13
c nmr(100mhz,cdcl3)δ144.01,128.71,127.84,127.03,86.70,72.26,70.63,63.36,61.93.
[0096]
第二步、单三苯甲基保护的六聚乙二醇的制备
[0097]
氮气氛围,0℃条件下,将7.4ml四氢呋喃和156mg(3.9mmol,1.5eq)氢化钠依次加入反应瓶,然后加入905.2mg(5.2mmol,1.0eq)单三苯甲基保护聚乙二醇/thf(5ml)溶液。保持0℃15分钟后逐滴滴入999.3mg(7.8mmol,1.5eq)四聚乙二醇磺酸内酯/四氢呋喃(3ml)溶液。滴加完毕后,0℃持续15min后升至室温。5h后tlc监测,待原料反应完毕后,氮气条件下依次慢慢加入0.1ml(5.2mmol,2.0eq)水和0.14ml(2.6mmol,1.0eq)硫酸(98%)。搅拌5mintlc监测反应完毕,向反应体系中慢慢滴加碳酸氢钠淬灭反应,二氯甲烷萃取三次,有机相用无水硫酸钠干燥,过滤,旋干,柱层析得产物1.0095g,产率74.1%,高效液相色谱(hplc)检测产品纯度为98%。1h nmr(400mhz,cdcl3)δ7.463(d,j=7.2hz,6h),7.224-7.290(s,9h),3.599-3.682(m,22h),3.226(s,2h).
13
c nmr(100mhz,cdcl3)δ144.13,128.73,127.80,126.95,86.52,72.55,70.79,70.72,70.69,70.62,70.56,70.33,63.32,61.76.
[0098]
第三步、三苯甲基保护的六聚乙二醇乙酸叔丁酯的制备
[0099]
在0℃,氮气条件下,依次向反应瓶中加入2.5682g(4.9mmol,1.0eq)单三苯甲基保护聚乙二醇,10ml dmf和294mg(1.5mmol,1.5eq)氢化钠。10min后向体系中逐滴滴加0.83ml(5.2mmol,1.05eq)/2-溴乙酸叔丁酯,0℃持续10分钟后升至室温并搅拌过夜。点板检测点板检测,待反应完毕后用水淬灭,乙酸乙酯萃取三次,有机相用饱和食盐水洗,合并有机相用无水硫酸钠干燥,过滤,旋干,柱层析得产物2.8087g,产率89.8%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ7.462(s,6h),7.236-7.286(m,9h),3.641-3.679(m,24h),3.235(s,2h),1.471(s,9h).
13
c nmr(100mhz,cdcl3)δ169.72,144.13,128.73,127.80,126.95,86.53,81.59,70.80,70.72,70.69,70.61,69.04,63.32,29.75,28.14.
[0100]
第四步、一端为游离羟基的六聚乙二醇乙酸叔丁酯的制备
[0101]
氮气条件下,将1.5320g(2.4mmol,1.0eq)三苯甲基保护聚乙二醇乙酸叔丁酯溶解到meoh/thf=1/1(6ml/6ml)混合溶液,然后再向混合液中加入45.7mg(0.24mmol,0.1eq)对甲苯磺酸水合物,并在30℃反应2.5h。点板检测点板检测反应完毕,碳酸氢钠淬灭后再在减压条件下旋去溶剂,残留物dcm萃取三次,合并有机相无水硫酸钠干燥,过滤旋干,柱层析得产物750.6mg,产率79%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ4.027(s,2h),3.612-3.710(m,24h),1.475(s,9h).
13
cnmr(100m hz,cdcl3)δ169.75,81.60,72.67,70.72,70.57,70.27,69.03,61.73,28.14.
[0102]
第五步、对甲苯磺酰氧基-六聚乙二醇-乙酸叔丁酯的制备
[0103]
氮气条件下,依次向圆底烧瓶加入750.6mg(1.9mmol,1.0eq)一端为游离羟基聚乙二醇乙酸叔丁酯、46.4mg(0.38mmol,0.2eq)dmap、4.5ml dcm和0.4ml(0.38mmol,1.5eq)三乙胺,然后在0℃条件下逐滴加入434.8mg(2.28mmol,1.2eq)对甲苯磺酰氯/dcm(1.0ml)溶液,滴加完毕后保持该温度反应10min后升至室温并搅拌过夜。tlc检测反应完毕,nh4cl淬灭,dcm萃取三次,有机相饱和食盐水洗,合并有机相用无水硫酸钠干燥,过滤旋干,柱层析得产物943mg,产率90.2%,高效液相色谱(hplc)检测产品纯度98%。1h nmr(400mhz,cdcl3)δ7.804(s,2h),7.351(s,2h),4.022(s,2h),3.586-3.695(m,24h),2.452(s,3h),1.474(s,9h).
13
c nmr(100mhz,cdcl3)δ169.72,144.84,132.93,129.86,128.03,81.60,70.76,70.72,70.61,70.57,69.28,69.03,68.70,28.14,21.70.
[0104]
根据上述说明书的揭示,本发明所属领域的技术人员还可以对上述实施方式进行适当的变更和修改。因此,本发明并不局限于上面揭示和描述的具体实施方式,对本发明的一些修改和变更也应当落入本发明的权利要求的保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
