用于复发型多发性硬化症(rms)的治疗性酪氨酸激酶抑制剂
1.本技术书主张2020年1月20日申请的美国临时申请no.62/963,238,2020年2月5日申请的美国临时申请no.62/970,502及2020年4月20日申请的美国临时申请案号63/013,895的优先权,其各自内容以引用的方式就所有目的并入本文中。
技术领域
2.本文涉及治疗性酪氨酸激酶抑制剂,尤其是布鲁顿酪氨酸激酶(“btk”)抑制剂用于治疗复发型多发性硬化症(rms)的领域。
背景技术:
3.多发性硬化症(ms)是一种影响全世界超过1百万人的神经性疾病。其为造成青年和中年人神经失能最常见的因素且对于患者和其家庭具有重要的身体、心理、社会和财务影响。ms涉及免疫介导的过程,该过程中身体免疫系统的异常反应针对中枢神经系统(cns)。在病程中,硬化,亦即病灶或瘢痕出现在神经细胞的髓鞘中,破坏电子信号的传递。硬化随时间累积并造成ms病患所经历的衰弱征候。ms病患一般会经历其各自可能为轻度、中度或严重的四个临床病程之一:临床单一综合征、复发缓解、续发性进展和原发性进展。大约85%的ms病患具有反复发作型的疾病,其中病患经历神经功能急性恶化情节的明确定义的复发(也称为爆发或恶化),接着是无疾病进展的部分或完全恢复期(缓解)。在本文的范围内,“复发型多发性硬化症”、“复发型ms”或“rms”可包括临床单一征候群(“cis”)、复发缓解硬化症(“rrms”)和复发续发性进展多发性硬化症(“r-spms”)。参见,例如lublin et al.,defining the clinical course of multiple sclerosis;the 2013 revisions,neurology 2014;83:278-286。
4.免疫调节药物是ms治疗的主流。最近的临床研究已验证以b淋巴细胞为目标的试剂,尤其是b细胞耗竭剂如奥瑞珠单抗(ocrelizumab)(抗-cd20)的效用(hauser et al.,nengl j med.2017;376(3):221-34)。瞄准b细胞代表着悖离以调节t细胞活性展现治疗利益的动物模型的主流教条并定位b细胞作为目前ms药物开发的核心(lehmann-horn k et al.,int j mol sci.2017;18(10):2048)。驻留在cns的免疫细胞的重要性亦已被熟知且在ms发病机制亦需考虑此项(hemmer b et al,nat clin pract neurol.2006;2(4):201-11)。
5.尽管有这些新近的进展,但对于以cns的神经发炎为目标,以停止患有复发型多发性硬化症(rms)和该疾病进展型(原发性进展型多发性硬化症(“ppms”)和非复发型续发性进展多发性硬化症(“nr-spms”))患者长期失能和神经退化的目的的治疗,仍有明显未满足的需求(stys pk et al,nat rev neurosci.2012;13(7):507-14)。即使最新近的高效改善疾病病程治疗也主要作用于外围的适应性免疫,仅具有中度或暂时的停止神经炎症和神经退化过程以及停止疾病进展的能力,还有在新近的进展型ms的研究中同样也验证了(montalban x et al,n engl j med.2017;376(3):209-20;kappos l et al,lancet 2018;391(10127):1263-73)。
6.尽管已批准的改善病程进展的治疗在防止急性复发上是有效的,但在现有的调节适应性免疫的细胞分子的策略之外,有越来越多的证据,由骨髓细胞株(骨髓衍生的单核细胞/巨噬细胞和cns-驻留微胶细胞)所介导的先天性免疫引起了许多持续的ms神经退化方面(hemmer b et al.,lancet neurol.2015;14(4):406-19;rahmanzadeh r et al.,rev neurosci.2018 jun 8)。针对先天性免疫的免疫调节具有削弱“郁积性发炎”和其他目前已批准治疗仍未解决的疾病进展表达的潜力。
7.布鲁顿氏酪氨酸激酶(btk)路径在b淋巴细胞和骨髓细胞(包括cns微胶细胞)的信号传导至关重要。各自这些细胞类型牵涉多发性硬化症(ms)的发病机制。再者,因为btk信号传递对于b细胞成熟成为分泌抗体血浆细胞至关重要,因此btk抑制可调节细胞免疫和体液免疫二者。因此,抑制btk信号传递的抑制剂代表瞄准二种免疫系统方面的双重机制。
技术实现要素:
8.因此,相较于目前可取得的治疗,抑制btk的化合物能抑制造成神经发炎的抗原引发性b细胞活化及调节连接脑中和脊髓中神经发炎的适应不良微胶细胞二者,可以较大利益有效的用于治疗rms。
9.因此,提供下列实施方案。在某些实施方案中,提供一治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供一降低新钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5一c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供一降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供一降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供在一患有复发型多发性硬化症(rms)的受试者中降低复发率的方法,该方法包括对该受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。
[0010]
在另外的实施方案中,btk抑制剂的剂量为大约5mg至约60mg。在另外的实施方案中,剂量为5mg。在另外的实施方案中,剂量为15mg。在另外的实施方案中,剂量为30mg。在另外的实施方案中,剂量为60mg。在某些实施方案中,治疗复发型多发性硬化症(rms)的方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中施用btk抑制剂,如mri所测,为抑制新的活动性脑病灶形成。在另外的实施方案中,施用该btk抑制剂化合物为作为单一治疗。在某些实施方案中,rms选自临床单一综合征(clinically isolated syndrome,cis)、复发缓解多发性硬化症(relapsing remitting multiple sclerosis,rrms)和复发续发性进展多发性硬化症(relapsing secondary progressive multiple sclerosis,r-spms)。在另外的实施方案中,该受试者为人类。
1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中在12周的btk给药后无新的gd-增强的t1高信号病灶形成。
[0023]
在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于治疗有需要受试者的复发型多发性硬化症(rms)的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有复发型多发性硬化症(rms)的受试者其新的或扩大的t2病灶数的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有复发型多发性硬化症(rms)的受试者其钆(gd)-增强的t1高信号病灶总数的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有多发性硬化症(ms)的受试者其复发率的方法中。
附图说明
[0024]
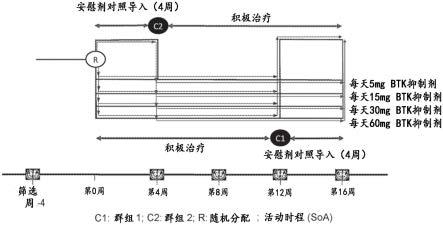
图1显示一示例性的整体治疗设计。
[0025]
图2a显示在12周的btk抑制剂治疗后,或第2群组病患在4周安慰剂治疗后,主要终点结果-新的gd-增强的t1高信号病灶数(第1群组:第12周;第2群组:第16周)。就基线gd-增强的t1高信号病灶活动性(有/无)使用负二项式模型调整病灶的相对降低数(rr)。ci:信赖区间。
[0026]
图2b显示在btk抑制剂治疗后,以多重比较法和模型化(mcp-mod)为基准的新的gd-增强的t1高信号脑病灶的估算的剂量反应曲线。选择最佳的拟合模型作为具有最小广义aic(赤池信息量准则)的模型。
[0027]
图3a显示在12周的btk抑制剂治疗或第2群组病患4周的安慰剂治疗后的次要终点结果-新的或扩大的t2病灶数(第1群组:第12周;第2群组:第16周)。就基线gd-增强的t2高信号病灶活动性(有/无)使用负二项式模型调整病灶的相对降低数(rr)。ci:信赖区间。
[0028]
图3b显示在btk抑制剂治疗后,以mcp-mod分析为基准新的或扩大的t2病灶数的估算的剂量反应曲线(次要终点)。
[0029]
图4显示在以igg治疗及以igg和btk抑制剂治疗后,相较于对照组,小鼠微胶细胞中差异表达基因(deg)的相对表达量。ctl=对照组;igg=免疫球蛋白;rgs1 g蛋白信号传递1的调节子。
[0030]
图5a至5b显示仅以igg和以igg与btk抑制剂体外治疗小鼠微胶细胞后(图5a),以及以btk抑制剂体外治疗初次受试小鼠的微胶细胞后(图5b),rgs1 mrna量化测量值。ctl=对照组;igg=免疫球蛋白;mrna=信使核糖核酸。
[0031]
图6显示以各种剂量的btk抑制剂(0.6、6和24mg/kg)治疗初次受试小鼠后,微胶细胞中的相对rgs1 mrna表达。veh:媒剂(对照组)。
[0032]
图7a显示以单一细胞rnaseq数据集为基准的umap(均匀流形逼近及投影)作图,其中辨识出各种cns细胞,包括微胶细胞(以空心圆表示)(图7a)。
[0033]
图7b显示在续发进展多发性硬化症(spms)病患和对照组中的相对rgs1量(图7b)。
具体实施方式
[0034]
现在将详细参照特定实施方案,其实施例伴随图式加以说明。在本文提供说明性实施方案的同时,应理解并不希望本发明受限于所述实施方案。相反,本发明意在涵盖可能包括在如所附的权利要求书定义的本文中的所有替代方案、修改和等价物。
[0035]
文中所用的章节标题仅作为组织目的且不应理解为在任何方面限制所欲的主题。如果以引用的方式所并入的任何文献与本说明书中所定义的任何术语相抵触时,以本说明书为准。在本发明结合各种实施方案说明的同时,不希望本发明被局限于这些实施方案。相反,本领域技术人员应能理解,本发明涵盖各种替代方案、修改和等价物。i.定义
[0036]
除非另有陈述,否则本说明书和权利要求书中所使用的下列术语就本文的目的加以定义并具有下列意义:
[0037]
如文中所用,“btk抑制剂”、“btk抑制剂化合物”和“该化合物”是指具有下列结构的(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮:该化合物也称为4-氨基-3-(4-苯氧基苯基)-1-[(3r)-1-(丙-2-烯酰基)哌啶-3-基]-1,3-二氢-2h-咪唑并[4,5-c]吡啶-2-酮,其具有下列结构:和/或其药学上可接受的盐。
[0038]“药学上可接受的载剂”或“药学上可接受的赋形剂”是指可用于制备药物组合物的载剂或赋形剂,其通常在生物上是安全、无毒及并非不合需求的,且包括兽医用途上以及人类药物用途上可接受的载剂和赋形剂。“药学上可接受的载剂/赋形剂”如本说明书和权利要求书中所用包括一种或一种以上的这些赋形剂。
[0039]“治疗”疾病包括:
(1)防止疾病,例如在可能暴露于或易罹患此疾病但尚未经历或展现此疾病征候的哺乳动物中不会发展出造成此疾病的临床征候;(2)抑制疾病,例如阻止或降低发生此疾病或其临床征候;或(3)减轻疾病,例如造成疾病或其临床征候消退。
[0040]“任选地”是指后续描述的事件或环境可能但不需要发生,且此叙述包括其中该事件或状况发生的情况及其中不发生的情况。
[0041]“治疗有效量”是指当施用哺乳动物供治疗一疾病时足以使此疾病的治疗产生效用的该btk抑制剂化合物的量。“治疗有效量”将依照化合物、疾病和其严重度以及所治疗的哺乳动物的年龄、体重等而变。
[0042]
在详细描述本发明之前,应理解本文不限于特定的组合物或方法步骤,因为其可能改变。
[0043]
应注意,如本说明书和所附的权利要求书中所用,除非文中明确指出,否则单数型“一”和“该”包括复数参照物。因此,例如,提到“一缀合物”包括复数个缀合物且提到“一细胞”包括复数个细胞及诸如此类。
[0044]
数字范围包括定义范围的数字。考虑有效数字和与测量有关的误差,测量的和可测量的值应理解为概约的。同样的,使用“包括”、“包含”、“含有”和“涵盖”不希望受限。应理解前述的一般说明和详细说明仅为示例性和说明性且并非限制本发明。
[0045]
除非在上述说明中特别注明,否则本说明书中实施方案陈述“包括”各种组份亦涵盖“由所述的组份组成”或“基本上由所述的组份组成”;而本说明书中实施方案陈述“基本上由各种组份组成”亦涵盖“包括”或“由所述的组份组成”或“包括”所述的组份(该可互换性并不适用于权利要求书中所使用的术语)。
[0046]
术语“其组合”如文中所用是指此术语前面所列出的术语的任何和所有的排列和组合。例如,“a、b、c或其组合”希望包括至少其中一项:a、b、c、ab、ac、bc或abc,且若顺序在特定的内容中为重要的,亦可为ba、ca、cb、acb、cba、bca、bac或cab。继续此实例,明确包含的为含有重复一个或多个项目的组合,例如bb、aaa、aab、bbc、aaabcccc、cbbaaa、cababb依此类推。本领域技术人员应理解,除非文中为显而易见的,否则典型地对于术语的数目或术语的任何组合并无限制。
[0047]
除非文中另有要求,否则“或”以包含在内的意义来使用,即等同于“和/或”。ii.施用的btk抑制剂化合物
[0048]
在某些实施方案中,施用btk抑制剂化合物(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮,供有需要的受试者来治疗复发型多发性硬化症(rms)。在某些实施方案中,该btk抑制剂化合物为(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的药学上可接受的盐。在某些实施方案中,施用治疗有效量的btk抑制剂化合物。在某些实施方案中,施用5至60mg剂量的btk抑制剂化合物。
[0049]
可根据例如美国专利第9,688,676 b2号中所述的方法和流程制备btk抑制剂化合物,尤其是第62行第8列至第65行第32列,及第67行第28列至第69行的内容,该内容以引用的方式并入本文中。
[0050]
给予下列(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并
[4,5-c]吡啶-2(3h)-酮化合物的制备使本领域技术人员能制备btk抑制剂化合物。该合成路径不应视为限制本文的范围,而仅仅是作为说明或其代表。
[0051]
示例性的(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的合成:
[0052]
于100ml圆底烧瓶中置入(r)-4-氨基-3-(4-苯氧基苯基)-1-(啶-3-基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮(150mg,0.37mmol,1.00当量)、dcm-ch3oh(6ml)、tea(113mg,1.12mmol,3.00当量)。接着于0℃搅拌下在5min内逐滴加入丙-2-烯酰氯(40.1mg,0.44mmol,1.20当量)。将生成的溶液于0℃搅拌2h。于真空下浓缩该生成的混合物。将残余物以二氯甲烷/甲醇(30:1)施用于硅胶柱。于下列条件下通过制备式-hplc纯化粗产物(100mg)(柱xbridge prep c
18 obd柱,5μm,19*150mm;移动相含0.05%tfa水和acn(25.0%can至高45.0%于8min内)。得到54.5mg的(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮产物为白色固体。lc-ms m/z:465.2(m 1)。iii.治疗方法
[0053]
文中提供治疗复发型多发性硬化症(rms)的方法,该方法包括施用有需要的受试者一治疗有效量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂化合物和/或其药学上可接受的盐。在某些实施方案中该治疗有效量为约5至约60mg。在某些实施方案中,该受试者为哺乳动物。在某些实施方案中,该哺乳动物为人类。在某些实施方案中,该受试者在治疗前具有一个或多个rms的征候且该治疗降低或消除该一个或多个征候。在某些实施方案中,该受试者患有神经病变疼痛、骨骼肌疼痛或rms所造成的痉挛。
[0054]
在某些实施方案中,一患有rms的受试者在过去1年内具有至少一次有记载的复发,和/或在过去2年内具有二次以上有记载的复发,和/或在过去6个月内和在筛选前经nmr扫描具有一个以上的活动性gd-增强脑病灶。
[0055]
在某些实施方案中,施用5至10mg、10至15mg、15至20mg、20至25mg、25至30mg、30至35mg、35至40mg、40至45mg、45至50mg、50至55mg或55至60mg的剂量。在某些实施方案中,该剂量为5mg、10mg、15mg、20mg、25mg、30mg、35mg、40mg、45mg、50mg、55mg或60mg。在某些实施方案中,该剂量为5mg。在某些实施方案中,该剂量为15mg。在某些实施方案中,该剂量为30mg。在某些实施方案中,该剂量为60mg。
[0056]
在某些实施方案中,每天给剂。每日剂量可以单一剂量或分成多个部分来递送。例如,在某些实施方案中,每天给剂一次(例如,大约每24小时)。在某些实施方案中,每天给剂二次。在某些实施方案中,该剂量为分成二个部分每天施用二次(例如,大约每12小时)。在某些实施方案中,该剂量为分成三个部分每天施用三次(例如,大约每8小时)。在某些实施方案中,该剂量为分成四个部分每天施用四次(例如,大约每6小时)。
[0057]
在某些实施方案中,以口服给剂。在某些实施方案中,以片剂的形式给剂。在某些实施方案中,以药片、胶囊、半固体、散剂、持续释放制剂、溶液、悬浮液、酏剂、气雾剂或任何其他适合的组合物形式给剂。
[0058]
在某些实施方案中,施用该受试者btk抑制剂化合物历时约4、8、12、16或20周的时间。在某些实施方案中,施用该受试者btk抑制剂化合物历时约12周的时间。在某些实施方案中,每天给剂一次。
[0059]
在某些实施方案中,随食物一起给剂。在某些实施方案中,随食物一起每天给剂一次。在某些实施方案中,随食物一起施用5mg、15mg、30mg或60mg的剂量。在某些实施方案中,随食物一起每天一次施用5mg、15mg、30mg或60mg的剂量。在某些实施方案中,随食物一起每天一次施用60mg的剂量。在某些实施方案中,以口服溶液或片剂来给剂。在某些实施方案中,以口服溶液或片剂随食物一起给剂。在某些实施方案中,以口服溶液或片剂每天给剂一次。在某些实施方案中,以口服溶液或片剂随食物一起每天给剂一次。在某些实施方案中,以口服溶液或片剂施用60mg的剂量。在某些实施方案中,以口服溶液或片剂随食物一起施用60mg的剂量。在某些实施方案中,以口服溶液或片剂每天施用60mg的剂量。在某些实施方案中,以口服溶液或片剂随食物一起每天一次施用60mg的剂量。
[0060]
rgs1(g蛋白信号传递1调节子)为作为g蛋白信号传递路径的负调节子且牵涉各种发炎性疾病。rgs1被鉴定为ms危险因子且亦发现富含于微胶细胞中(international multiple sclerosis genetics consortium,science 365:6460(2019))。如下文实例3中所详述的,rna定序显示小鼠微胶细胞中的btk-依赖的转录标签,及rgs1经鉴定在此btk-依赖的微胶细胞标签中为其中一个上调的基因。在实例3的体外和体内研究中进一步显示btk抑制作用使得被igg活化的小鼠微胶细胞标签正常化,包括下调rgs1。因此,在某些实施方案中,施用btk抑制剂降低了脑细胞中的rgs1表达。脑细胞可能包括微胶细胞。在某些实施方案中,于体外和体内测量脑细胞中的rgs1表达量。
[0061]
在某些实施方案中,施用btk抑制剂降低了新的活动性病灶。在某些实施方案中,施用btk抑制剂降低了新的活动性钆(gd)-增强的t1高信号病灶。在某些实施方案中,施用btk抑制剂降低了新的或扩大的t2病灶。
[0062]
在某些实施方案中,施用btk抑制剂,如mri所测,降低了新的钆(gd)-增强的t1高信号病灶。在某些实施方案中,新的gd-增强的t1高信号病灶的数目少于1。在某些实施方案中,新的gd-增强的t1高信号病灶数等于或少于0.77、0.7、0.6、0.5、0.4、0.3、0.2或0.1。在某些实施方案中,在12周的btk抑制剂治疗后无新的gd-增强的t1高信号病灶形成。
[0063]
在某些实施方案中,施用btk抑制剂,如mri所测,降低了新的或扩大的t2病灶数目。在某些实施方案中,新的或扩大的t2病灶数目等于或少于2。在某些实施方案中,新的或扩大的t2病灶数等于或少于1.9、1.8、1.7、1.6、1.5、1.4、1.3、1.2、1.1、1.0、0.9、0.8、0.7、0.6.0.5、0.4、0.3、0.2或0.1。在某些实施方案中,在12周的btk抑制剂治疗后无新的或扩大
的t2病灶形成。
[0064]
在某些实施方案中,施用btk抑制剂在12周的btk抑制剂治疗后,降低了gd-增强的t1高信号病灶总数。
[0065]
在某些实施方案中,剂量为60mg,且在12周的btk抑制剂治疗后形成1或0个新的gd-增强的t1高信号病灶。在某些实施方案中,在12周的btk抑制剂治疗后形成0个新的gd-增强的t1高信号病灶。在某些实施方案中,新的或扩大的t2病灶数等于或少于2。在某些实施方案中,新的或扩大的t2病灶数等于或少于2、1.9、1.8、1.7、1.6、1.5、1.4、1.3、1.2、1.1、1.0、0.9、0.8、0.7、0.6.0.5、0.4、0.3、0.2或0.1。
[0066]
在某些实施方案中,施用btk抑制剂,在12周的btk抑制剂治疗后降低了gd-增强的t1-高信号病灶总数。
[0067]
在一实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用60mg的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中在12周的btk抑制剂给药后,无新的gd-增强的t1高信号病灶形成。
[0068]
在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时约12周的时间。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。
[0069]
在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有
4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供一降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供一降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低新的钆(gd)-增强的t1高信号病灶数的受试者。
[0071]
在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数目的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的钆(gd)-增强的t1高信号病灶数的方法,该方法包括对患有复发型多发性硬化
症(rms)的受试者60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低新的钆(gd)-增强的t1高信号病灶数的受试者。
[0072]
在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供一降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供一降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低新的或扩大的t2病灶数的受试者。
[0073]
在某些实施方案中,提供一降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用30mg
包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低新的或扩大的t2病灶数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低新的或扩大的t2病灶数的受试者。
[0074]
在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一
次历时至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低钆(gd)-增强的t1高信号病灶总数的受试者。
[0075]
在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低钆(gd)-增强的t1高信号病灶总数的方法,该方法包括对患有复发型多发性硬化症(rms)的受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有复发型多发性硬化症(rms),需要降低钆(gd)-增强的t1高信号病灶总数的受试者。
[0076]
在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用包括(r)-1一(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有多发性硬化症,需要降低复发率的受试者。
[0077]
在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供一降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物施用至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有多发性硬化症,需要降低复发率的受试者。
[0078]
在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用5mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯
基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用15mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用30mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在某些实施方案中,提供降低患有多发性硬化症(ms)受试者的复发率的方法,该方法包括对该受试者施用60mg包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,其中该btk抑制剂化合物每天给药一次历时至少约12周的时间。在上述提供方法的某些实施方案中,该btk抑制剂施用患有多发性硬化症,需要降低复发率的受试者。
[0079]
在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于治疗有此需要受试者的复发型多发性硬化症(rms)的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有复发型多发性硬化症(rms)受试者其新的或扩大的t2病灶数的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有复发型多发性硬化症(rms)受试者其钆(gd)-增强的t1高信号病灶总数的方法中。在某些实施方案中,提供包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂,以用于降低患有多发性硬化症(ms)受试者的复发率的方法中。
[0080]
在某些实施方案中,该btk抑制剂化合物以单一疗法给药。在某些实施方案中,该方法包括施用该btk抑制剂化合物及至少一另外的治疗剂。该另外的治疗剂可与btk抑制剂化合物同时或先后给药。
[0081]
给药频率可由本领域技术人员,例如主治医师,以所欲治疗的症状、所欲治疗病患的年龄、所欲治疗症状的严重度、所欲治疗受试者的整体健康状况及诸如此类为基准来决定。在某些实施方案中,btk抑制剂化合物以用于治疗rms的治疗有效量来给药。该治疗有效量典型地依照所欲治疗受试者的体重、其身体或健康状况、所欲治疗症状的广泛性或所欲治疗受试者的年龄、药物调配方法和/或给药方法(例如,给药时间和给药路径)而定。
[0082]
在某些实施方案中,提供治疗复发型多发性硬化症(rms)的方法,该方法包括对有需要的受试者施用约5至约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。在某些实施方案中,施用该抑制剂降低了新的活动性脑病灶数。在某些实施方案中,该病灶为gd-增强的t1-高信号病灶。在某些实施方案中,该病灶数为通过核磁共振成像(mri)来检测。
[0083]
在某些实施方案中,提供一治疗rms的方法,该方法包括对有需要的受试者施用约5至10mg,10至15mg,15至20mg,20至25mg,25至30mg,30至35mg,35至40mg,40至45mg,45至
50mg,50至55mg或55至60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。在某些实施方案中,提供治疗rms的方法,该方法包括对有需要的受试者施用约5mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。在某些实施方案中,提供治疗rms的方法,该方法包括对有需要的受试者施用约15mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。在某些实施方案中,提供治疗rms的方法,该方法包括对有需要的受试者施用约30mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。在某些实施方案中,提供治疗rms的方法,该方法包括对有需要的受试者施用约60mg剂量的包括(r)-1-(1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1h-咪唑并[4,5-c]吡啶-2(3h)-酮的btk抑制剂和/或其药学上可接受的盐。
[0084]
制剂的选择依照各种因素而定,例如药物给药模式(例如,就口服给药,片剂、药片或胶囊形式的制剂为优选)及药物的生物利用度。最近,依据通过增加表面积,亦即降低粒子大小能增加生物利用度的原则为基础,已开发出特别可用于显现出低生物利用度的药物的药物制剂。例如,美国专利第4,107,288号描述了具有粒子大小范围从10至1,000nm的药物制剂,其中该活性物质为承载在一交联的大分子基质上。美国专利第5,145,684号描述了一药物制剂的制造,其中该药物在表面修饰剂的存在下粉末化至纳米粒子(400nm的平均粒大小),及然后分散于液体媒剂中,得到展现显著高生物利用度的药物制剂。在胃ph分解的药物的生物利用度可通过以十二指肠中释放药物的制剂施用该药物来增加。
[0085]
组合物一般由btk抑制剂化合物和/或其药学上可接受的盐与一药学上可接受赋形剂,例如帮助该btk抑制剂化合物和/或其药学上可接受的盐加工处理成为可药物上使用的制剂的粘结剂、表面活性剂、稀释剂、缓冲剂、抗黏附剂、助流剂、亲水或疏水性聚合物、阻燃剂、稳定剂、崩解剂或超级崩解剂、消泡剂、填充剂、风味剂、色剂、润滑剂、吸附剂、防腐剂、塑化剂或甜味剂或其混合物组合所组成。只要适合且为本领域所知,可使用任何熟知的技术或赋形剂,参见,例如remington:the science and practiceof pharmacy,twenty-first ed.,(pharmaceutical press,2005);liberman,h.a.,lachman,l.,and schwartz,j.b.eds.,pharmaceutical dosage forms,vol.1-2taylor&francis 1990;及r.i.mahato,ansel’s pharmaceutical dosage forms and drug delivery systems,second ed.(taylor&francis,2012).
[0086]
在特定的实施方案中,制剂可包括一种或多种ph调节剂或缓冲剂,例如酸类,如乙酸、硼酸、柠檬酸、延胡索酸、马来酸、酒石酸、苹果酸、乳酸、磷酸和盐酸;碱类,如氢氧化钠、磷酸钠、硼酸钠、柠檬酸钠、乙酸钠、乳酸钠和三-羟甲基氨基甲烷;以及缓冲剂,如柠檬酸盐/右旋糖、碳酸氢钠、氯化铵及诸如此类。这些用作为碱的缓冲剂可能具有钠以外的其他相反离子,例如钾、镁、钙、铵或其他相反离子。这些酸、碱和缓冲剂以维持组合物ph在一可接受范围内所需的量纳入。
[0087]
在特定的实施方案中,制剂亦可包括一种或多种使组合物的渗透压达到可接受范围所需的量的盐类。这些盐类包括所述具有钠、钾或铵阳离子及氯化物、柠檬酸盐、抗坏血
酸盐、硼酸盐、磷酸盐、碳酸氢盐、硫酸盐、硫代硫酸盐或硫酸氢盐阳离子的盐类;适合的盐类包括氯化钠、氯化钾、硫代硫酸钠、硫酸氢钠和硫酸铵。
[0088]
在特定的实施方案中,制剂亦可包括一种或多种消泡剂用以降低可能造成水性分散液凝结,成膜中起泡或通常影响加工的加工期间的起泡。示例性的消泡剂包括硅乳化剂或山梨醇酐倍半油酸酯。
[0089]
在特定的实施方案中,制剂亦可包括一种或多种抗氧化剂,例如非硫醇抗氧化剂,例如二丁基羟基甲苯(bht)、抗坏血酸钠、抗坏血酸或其衍生物,以及生育酚或其衍生物。在特定的实施方案中,抗氧化剂在需要时增进化学稳定性。亦可加入其他的试剂,例如柠檬酸或柠檬酸盐或edta用以推迟氧化。
[0090]
在特定的实施方案中,制剂亦可包括一种或多种防腐剂用以抑制微生物活性。适合的防腐剂包括含汞物质,例如硝酸苯汞(merfen)和硫柳汞(thiomersal);稳定性二氧化氯;和季铵化合物,例如苯扎氯铵(benzalkonium chloride)、溴化十六烷基三甲铵和氯化十六烷基吡片。
[0091]
在特定的实施方案中,制剂亦可包括一种或多种粘结剂。粘结剂为赋予黏附性质并包括,例如海藻酸及其盐类;纤维素衍生物,例如羧甲基纤维素、甲基纤维素(例如)、羟丙基甲基纤维素、羟乙基纤维素、羟丙基纤维素(例如)、乙基纤维素(例如),以及微晶纤维物(例如);微晶右旋糖;直链淀粉;硅酸镁铝;多糖酸;澎润土;明胶;聚乙烯吡咯烷酮/乙酸乙烯酯共聚物;交链聚维酮;聚维酮;淀粉;预明胶化淀粉;黄蓍胶、糊精、糖,例如蔗糖(例如)、葡萄糖、右旋糖、糖蜜、甘露醇、山梨醇、木糖醇(例如)和乳糖;天然或合成胶,例如阿拉伯胶、黄蓍胶、甘地胶芒麻胶、聚乙烯吡咯烷酮(例如cl、cl、xl-10)、落叶松阿拉伯半乳聚糖、聚乙二醇、聚环氧乙烷、蜡、海藻酸钠及诸如此类。
[0092]
在特定的实施方案中,制剂亦可包括一种或多种分散剂和/或黏度调节剂。分散剂和/或黏度调节剂包括通过液体媒剂控制药物的扩散和均质度或造粒的方法或混拌方法。在某些实施方案中,这些试剂亦促进包膜或溶蚀基质的效能。示例性的扩散促进剂/分散剂包括,例如亲水性聚合物、电解质、60或80、peg、聚乙烯吡咯烷酮(pvp;商品名)及碳水化合物为基底的分散剂,例如,举例而言羟丙基纤维素(例如,hpc、h
‑‑
pc-sl和hpc-l)、羟丙基甲基纤维素(例如,hpmc k100、rpmc k4m、hpmc k15m、and hpmc k100m)、羧甲基纤维素钠、甲基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素邻苯二甲酸酯、羟丙基-甲基纤维素乙酸硬脂酸酯(hpmcas)、非晶纤维素、聚环氧乙烷、硅酸镁铝、三乙醇胺、聚乙烯醇(pva)、乙烯吡咯烷酮/乙酸乙烯酯共聚物(s630)、4-(1,1,3,3-四甲基丁基)-酚与环氧乙烷和甲醛的聚合物(也称为泰洛沙泊(tyloxapol))、泊洛沙姆(poloxamer)(例如pluronics和8,为环氧乙烷和环氧丙烷的嵌段共聚物);和泊洛沙明(poloxamine)(例如,tetronic也称为poloxamine为衍生自环氧丙烷和环氧乙烷逐步加成至乙二胺的四功能嵌段共聚物(basf corporation,parsippany,n.j.))、聚乙烯吡咯烷酮k12、聚乙烯吡咯烷酮k17、聚乙烯吡咯烷酮k25或聚乙
烯吡咯烷酮k30、聚乙烯吡咯烷酮/乙酸乙烯酯共聚物(s-630)、聚乙二醇,例如该聚乙二醇可具有约300至约6000,或约3350至约4000,或约7000至5400的分子量,羧甲基纤维素钠、甲基纤维素、聚山梨醇酯-80、海藻酸钠、胶例如黄蓍胶和阿拉伯胶、瓜尔豆胶、黄原胶,包括黄原胶、糖类、纤维素,例如羧甲基纤维素钠、甲基纤维素、羧甲基纤维素钠、聚山梨醇酯-80、海藻酸钠、聚乙氧山梨醇酐单月桂酸酯、聚维酮(povidone)、卡波姆(carbomer)、聚乙烯醇(pva)、海藻酸盐、几丁聚糖及其混合物。塑化剂,例如纤维素或三乙基纤维素亦可用做分散剂。特别可用于脂质体分散液和自乳化分散液中的分散剂有二肉豆蔻酰基磷脂酰胆碱、来自蛋类的天然磷脂酰胆碱、胆固醇和肉荳蔻酸异丙酯。一般而言,在填充粉末的明胶胶囊调配中使用约10至约70%的粘结剂量。片剂制剂中粘结剂的用量依照是否直接压制、湿式造粒、辗压或使用其他赋形剂,例如其本身可作为适度粘结剂的填充剂。具有本领域技术的制剂者可决定用于制剂的粘结剂的量,但片剂制剂中至高90%及更典型地至高70%的粘结剂用量是常见的。
[0093]
在特定的实施方案中,制剂亦可包括一种或多种有关在递送前用于稀释感兴趣化合物的化学化合物的稀释剂。稀释剂亦可用于稳定化合物,因为稀释剂可提供更稳定的环境。在本领域中利用溶于缓冲溶液中的盐类(其亦可提供ph控制和维持)作为稀释剂,其包括但不限于磷酸盐缓冲食盐水溶液。在特定的实施方案中,稀释剂为增加组合物的量,用以帮助压制或制造足够的量供作为胶囊填充的均匀混拌物。这些化合物包括,例如乳糖、淀粉、甘露醇、山梨醇、右旋糖、微晶纤维素例如磷酸氢钙、磷酸氢二水合物;磷酸钙;无水乳糖、喷雾干燥乳糖;预明胶化淀粉、可压缩糖,例如(amstar);羟丙基-甲基纤维素、羟丙基甲基纤维素乙酸硬脂酸酯、蔗糖为基底的稀释剂、糖粉;硫酸二氢钙单水合物、硫酸钙二水合物;乳酸钙三水合物、葡萄糖结合剂;水解谷类固体、直链淀粉;纤维素粉、碳酸钙;甘油、高岭土;甘露醇、氯化钠;肌醇、澎润土及诸如此类。
[0094]
在特定的实施方案中,制剂亦可包括一种或多种崩解剂,其包括当与胃肠液接触时,溶解和分散剂型二者。崩解剂为帮助破坏或崩解物质。崩解剂的实例包括淀粉,例如天然淀粉,例如玉米淀粉或马铃薯淀粉、预明胶化淀粉,例如national 1551或甘醇酸淀粉钠如或纤维素例如木材产品、甲基晶体纤维素,例如ph101、ph102、ph105、p100、和甲基纤维素、交联羧甲纤维素(croscarmellose)或交联纤维素,例如交联羧甲基-纤维素钠交联羧甲基纤维素或交联交联羧甲纤维素,交联淀粉,例如甘醇酸淀粉钠、交联聚合物,例如交联聚维酮、交联聚乙烯吡咯烷酮,海藻酸盐,例如海藻酸或海藻酸盐,例如海藻酸钠,黏土,例如hv(硅酸镁铝),胶例如琼脂、瓜尔胶、刺槐豆胶、卡拉胶(karaya)、果胶或黄蓍胶、甘醇酸淀粉钠、澎润土、天然海绵、表面活性剂、树脂例如阳离子交换树脂、柑橘渣、月桂基硫酸钠、月桂基硫酸钠与淀粉组合及诸如此类。
[0095]
在特定的实施方案中,制剂亦可包括一种或多种溶蚀促进剂。溶蚀促进剂包括在胃肠液中控制溶蚀特定物质的物质。溶蚀促进剂一般已为本领域的一般技术者所知。示例性的溶蚀促进剂包括,例如亲水性聚合物、电解质、蛋白、肽和氨基酸。
[0096]
在特定的实施方案中,制剂亦可包括一种或多种填充剂,其包括例如乳糖、碳酸
钙、磷酸钙、磷酸氢钙、硫酸钙、微晶纤维素、纤维素粉、右旋糖、葡萄糖结合剂、葡聚糖、淀粉、明胶化淀粉、蔗糖、木糖醇、乳糖醇、甘露醇、山梨醇、氯化钠、聚已二醇及其类似物。
[0097]
在特定的实施方案中,制剂亦可包括一种或多种调味剂和/或甜味剂,例如阿拉伯胶糖浆、乙酰磺氨酸钾(acesulfame k)、阿利甜(alitame)、大茴香、苹果、阿斯巴甜(aspartame)、香蕉、巴伐利亚奶油莓果、黑醋栗、奶油糖、柠檬酸钙、樟脑、焦糖、樱桃、樱桃奶油巧克力、肉桂、口香糖、柑橘、柑橘汽水、柑橘奶油、棉花糖、可可、可乐、酷樱桃(cool cherry)、酷柑橘(cool citrus)、赛克拉美(cyclamate)、右旋糖、尤加利、丁香酚、果糖、水果饮品、姜、甘草酸(glycyrrhizinate)、甘草糖浆、葡萄、葡萄柚、蜂蜜、异麦芽酮糖醇(isomalt)、柠檬、莱姆、柠檬奶油、甘草酸单铵、麦芽酚、甘露醇、枫糖、棉花糖、薄荷脑、薄荷奶油、混合莓果、新橙皮苷二氢查耳酮(neohesperidine dc)、纽甜(neotame)、橙皮、桃子、椒薄荷、椒薄荷奶油、粉、小红莓、根汁、朗姆酒、糖精、黄樟油精、山梨醇、绿薄荷、绿薄荷奶油、草莓、草莓奶油、甜菊糖、三氯蔗糖、蔗糖、糖精钠、糖精、阿斯巴甜、乙酰磺氨酸钾、甘露醇、踝蛋白(talin)、木糖醇、三氯蔗糖、山梨醇、瑞士奶油(swiss cream)、塔格糖(tagatose)、橘子、索马甜(thaumatin)、水果丁糖(tutti frutti)、香草、核桃、西瓜、野莓、冬青、木糖醇或这些调味成份的任何组合,例如大茴香-薄荷脑、樱桃-大茴香、肉桂-柳橙、樱桃-肉桂、巧克力-薄荷、蜂蜜-柠檬、柠檬-莱姆、薄荷脑-尤加利、柳橙-奶油、香草-薄荷及其混合物。
[0098]
在特定的实施方案中,制剂亦可包括一种或多种润滑剂和助流剂,其为防止、降低或抑制物质黏附或摩擦的化合物。示例性的润滑剂包括,例如硬脂酸、氢氧化钙、滑石、硬脂酰延胡索酸钠(sodium stearyl lumerate),烃,例如矿物油,或氢化植物油,例如氢化大豆油、高脂肪酸及其碱金属和碱土金属盐类,例如铝、钙、镁、锌,硬脂酸、硬脂酸钠、甘油、滑石、蜡、硼酸、苯甲酸钠、乙酸钠、氯化钠、亮氨酸、聚乙二醇(例如peg4000)或甲氧基聚乙二醇,例如油酸钠、苯甲酸钠、甘油二十二烷酸酯、聚乙二醇、月桂基硫酸镁或月桂基硫酸钠、胶体氧化硅,例如淀粉,例如玉米淀粉、硅油,表面活性剂及诸如此类。
[0099]
在特定的实施方案中,制剂亦可包括一种或多种塑化剂,其为用于软化肠衣或延迟释放膜衣使其较不易碎的化合物。适合的塑化剂包括,例如聚乙二醇,例如peg 300、peg 400、peg 600、peg 1450、peg 3350和peg 800,硬脂酸、丙二醇、油酸、柠檬酸三乙酯、癸二酸二丁酯、三乙基纤维素和三乙酸甘油酯。在某些实施方案中,塑化剂亦可作为分散剂或湿润剂。
[0100]
在特定的实施方案中,制剂亦可包括一种或多种稳定剂,其包括例如三乙酸甘油酯、三乙基柠檬酸酯、油酸乙酯、辛酸乙酯、月桂基硫酸钠、多库酯钠(sodium doccusate)、维生素e tpgs、二甲基乙酰胺、n-甲基吡咯烷酮、n-羟乙基吡咯烷酮、聚乙烯吡咯烷酮、羟丙基甲基纤维素、羟丙基环糊精,例如乙醇、正丁、异丙醇、胆固醇、胆盐、聚乙二醇200-600、四氢呋喃聚乙二醇醚(glycofurol)、transcutol、丙二醇和异山梨醇二甲醚及诸如此类的化合物。在一实施方案中,该稳定剂为维生素e tpgs和/或或β-羟丙基环糊精。
[0101]
在特定的实施方案中,制剂亦可包括一种或多种悬浮剂,其包括例如聚乙烯吡咯烷酮,例如聚乙烯吡咯烷酮k112、聚乙烯吡咯烷酮k17、聚乙烯吡咯烷酮k25或聚乙烯吡咯烷酮k30、乙烯吡咯烷酮/乙酸乙烯酯共聚物(s630)、聚乙二醇,例如该聚乙二醇可具有约300至约6000,或约3350至约4000,或约7000至约5400的分子量,羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、羟甲基纤维素乙酸硬脂酸酯、聚山梨醇酯-80、羟乙基纤维素、海藻酸钠,胶例如黄耆胶和阿拉伯胶、瓜尔豆胶、黄原胶,包括黄原胶、糖类,纤维素,例如羧甲基纤维素钠、甲基纤维素、羧甲基纤维素钠、羟丙基甲基纤维素、羟乙基纤维素、聚山梨醇酯-80、海藻酸钠、聚乙氧基山梨醇酐单月桂酸酯、聚乙氧基山梨醇酐单油酸酯、聚维酮及诸如此类的化合物。
[0102]
在特定的实施方案中,制剂亦可包括一种或多种表面活性剂,其包括例如月桂基硫酸钠、多库酯钠tween20、60或80、三乙酸甘油酯、维生素e tpgs、山梨醇酐单油酸酯、聚氧乙烯山梨醇酐单油酸酯、聚乙氧基山梨醇酐单月桂酸酯、聚山梨醇酯、胆盐、甘油基单硬脂酸酯、环氧乙烷和环氧丙烷的共聚物,例如(basf)及诸如此类的化合物。一些其他的表面活性剂包括聚氧乙烯脂肪酸甘油酯及植物油,例如聚氧乙烯(60)氢化蓖麻油;和聚氧乙烯烷基醚和烷基苯基醚,例如辛基酚聚醚10、辛基酚聚醚40。在某些实施方案中,可包括表面活性剂用以增进物理稳定性或用于其他目的。
[0103]
在特定的实施方案中,制剂亦可包括一种或多种增黏剂,其包括,例如甲基纤维素、黄原胶、羧甲基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羟丙基甲基纤维素乙酸硬脂酸酯、羟丙基甲基纤维素邻苯二甲酸酯、卡波姆(carbomer)、聚乙烯醇海藻酸酯、阿拉伯胶、几丁聚糖及其组合物。
[0104]
在特定的实施方案中,制剂亦可包括一种或多种湿润剂,其包括例如油酸、单硬脂酸甘油酯、山梨醇酐单油酸酯、山梨醇酐单月桂酸酯、三乙醇胺油酸酯、聚氧乙烯山梨醇酐单油酸酯、聚氧乙烯山梨醇酐单月桂酸酯、多库酯钠、油酸钠、月桂基硫酸钠、多库酯钠、三乙酸甘油酯、tween 80、维生素e tpgs、铵盐及诸如此类的化合物。
[0105]
文中所公开的药物制剂可通过将一种或多种固体赋形剂,例如载剂、粘结剂、填充剂、悬浮剂、调味剂、甜味剂、崩解剂、分散剂、表面活性剂、润滑剂、色剂、稀释剂、增溶剂、润湿剂、塑化剂、稳定剂、渗透增强剂、湿润剂、消泡剂、抗氧化剂、防腐剂或一种或多种其与一种或多种文中所述的化合物组合混合来获得,任选地将所生成的混合物研磨,若需要并在添加适合的赋形剂后,将颗粒混合物加工,得到片剂。
[0106]
文中所公开的药物制剂亦包括明胶制成的胶囊,以及明胶和塑化剂,例如甘油或山梨醇制成的软式封闭胶囊。胶囊亦可由聚合物例如羟丙基甲基纤维素(hypromellose)制成。胶囊可含有活性成份混合填充剂例如乳糖,粘结剂例如淀粉,和/或润滑剂例如滑石或硬脂酸镁,以及任选地稳定剂的混合物。在软式胶囊中,活性化合物可溶解活悬浮于液体中,例如油脂、液态石蜡、脂质、增溶剂或液态聚乙二醇。此外,可加入稳定剂。所有用于口服给药的制剂应为适合此给药的剂型。
[0107]
这些制剂可通过常规的药理学技术来制造。常规的药理学技术包括,例如一下列方法或其组合:(1)干混,(2)直接压制,(3)研磨,(4)干式或非水性造粒,(5)湿式造粒,(6)融合,或(7)挤压。参见,例如lachman et al.,the theory and practice of industrial pharmacy,3
rd ed.(1986)。其他的方法包括,例如喷雾干燥、锅包衣、熔化造粒、造粒、流化床
喷雾干燥或包衣[例如沃斯特包衣(wurstercoating)]、正切包衣、上方喷雾、制片、挤压、挤压/滚圆,及诸如此类。
[0108]
应理解,用于文中所述的固体剂型中的赋形剂间有大量的重叠。因此,上述所列的添加物仅仅作为示例且并非限制可包括在文中所述的固体剂型中的赋形剂类型。此赋形剂的类型和量可由本领域技术人员根据所述的特定性质,轻易决定。
[0109]
在某些实施方案中,文中所述的固体剂型可为包覆肠衣口服剂型,亦即文中所述的药物组合物的口服剂型,利用肠衣使化合物在胃肠道的肠中释放。“肠衣”药物和/或片剂是指包覆上在胃中保持完整但一旦到达肠中(在一实施方案中为小肠)则溶解和释放该药物的物质的药物和/或片剂。如文中所用“肠衣”为一种物质,例如一种聚合物物质或包住作为剂型或颗粒的治疗活性剂核心的物质。典型地,实质的量或所有肠衣物质在治疗活性剂从剂型释放之前溶解,以便于在小肠和/或大肠中达到延迟溶解治疗活性剂核心或颗粒。肠衣论述于,例如loyd,v.allen,remington:the science and practice of pharmacy,twenty-first ed.,(pharmaceutical press,2005;及p.j.tarcha,polymers for controlled drug delivery,chapter 3,crc press,1991中。于药物组合物上涂覆肠衣的方法已为本领域所熟知,并包括例如美国专利公开案第2006/0045822号。
[0110]
包覆肠溶膜衣的剂型可为含有btk抑制剂化合物和/或其药学上可接受的盐和/或其他赋形剂的颗粒、粉末、丸团、小珠或粒子的压制或模造或挤压片剂(包衣或未包衣),其中该颗粒、粉末、丸团、小珠或粒子本身为包膜或未包膜的,其限制条件为至少该片剂或btk抑制剂化合物为包膜的。肠衣口服剂型亦可为含有btk抑制剂化合物和/或其药学上可接受的盐和/或其他赋形剂的丸团、小珠或颗粒的胶囊(包膜或未包膜),其中该丸团、小珠或颗粒本身为包膜或未包膜的,但限制条件为至少其中一样为包膜的。一些原本用作为肠溶膜衣的膜衣的实例有蜂蜡和甘油单硬脂酸酯;蜂蜡、虫胶和纤维素;及鲸蜡醇、乳香和虫胶以及虫胶和硬脂酸(美国专利第2,809,918号);聚乙烯乙酸酯和乙基纤维素(美国专利第3,835,221号)。更新近,所用的膜衣为聚甲基丙烯酸酯的中性共聚物(eudragit l30d)(f.w.goodhart et al,pharm.tech.,p.64-71,april,1984);甲基丙烯酸和甲基丙烯酸甲酯的共聚物(eudragit s),或含有硬脂酸金属盐的聚甲基丙烯酸酯的中性共聚物(mehta等人美国专利第4,728,512号和第4,794,001号),纤维素乙酸琥珀酸酯及羟丙基甲基纤维素邻苯二甲酸酯。
[0111]
任何展现ph依赖性的溶解样貌的阴离子聚合物可在文中所述的方法和组合物中用作为肠溶膜衣,达成递送至肠中。在一实施方案中,递送可至小肠中。在另外的实施方案中,递送可至十二指肠中。在某些实施方案中,文中所述的聚合物为阴离子羧基聚合物。在其他的实施方案中,聚合物和其兼容的混合物,及某些其属性,包括(但不限于):
[0112]
虫胶:也称为纯化虫胶,其为得自昆虫的树脂状分泌物的精制产品。此膜衣溶于ph>7的媒剂中;
[0113]
丙烯酸聚合物:丙烯酸聚合物的效能(主要为其在生物流体中的溶解度)依照取代的程度和类型,可能不同。适合的丙烯酸聚合物的实例包括甲基丙烯酸共聚物和甲基丙烯酸铵共聚物。可取得的eudragit l、s和rs系列(由rohm pharma制造及已知为)为溶解于有机溶剂,水性分散液或干粉剂。eudragit l、s和rs系列不溶于胃肠道但可穿透且主要为用作结肠靶向。eudragit l、l-30d和s系列不溶于胃但溶于肠中且可经选择和调配
于大于5.5或低于5或大于7的ph值下溶解;
[0114]
纤维素衍生物:适合的纤维素衍生物的实例有:乙基纤维素;部分的纤维素的乙酸酯与邻苯二甲酸酐的反应混合物。依照取代的程度和类型,效能可能不同。纤维素乙酸苯甲酸酯(cap)在ph>6溶解。水三联(aquateric)(fmc)为水性基底系统且具有粒子<1μm的喷雾干燥cap拟乳胶(pseudolatex)。水三联(aquateric)中的其他组份可包括伯洛沙姆(pluronic)、tweens和乙酰基单甘油酯。其他的适合纤维素衍生物包括;纤维素乙酸偏苯三酸酯(eastman);甲基纤维素(pharmacoat,methocel);羟丙基甲基纤维素邻苯二甲酸酯(hpmcp);羟丙基甲基纤维素琥珀酸酯(hpmcs);和羟丙基甲基纤维素乙酸琥珀酸酯(hpmcas例如,aqoat(shinetsu))。依照取代的程度和类型,可能不同。例如hpmcp,例如hp-50、hp-55、hp-55s、hp-55f等级为适合的。依照取代的程度和类型,可能不同。例如适合等级的羟丙基甲基纤维素乙酸琥珀酸酯包括,但不限于as-lg(lf),其于ph 5时溶解,as-mg(mf)于ph 5.5时溶解,及as-hg(hf)于较高的ph溶解。这些聚合物以颗粒或细粉提供,供用于水性分散液;
[0115]
聚乙烯乙酸邻苯二甲酸酯(pvap):pvap溶于ph>5,且对水蒸气和胃液的穿透力极低。上述聚合物的详细说明及其ph-依赖的溶解度可参见professor karl thoma和karoline bechtold于http://pop.www.capsugel.com/media/library/enteric-coated-hard-gelatin-capsules.pdf上,标题“enteric coated hard gelatin capsules”的文献。在某些实施方案中,膜衣可能且通常真正含有塑化剂及可能含有其他膜衣赋形剂,例如本领域中熟知的色剂、滑石和/或硬脂酸镁。适合的塑化剂包括柠檬酸三乙酯(citroflex 2)、三乙酸甘油酯、乙酰柠檬酸三乙酯(citroflec a2)、carbowax 400(聚乙二醇400)、邻苯二甲酸二乙酯、柠檬酸三丁酯、乙酰单甘油酯、甘油、脂肪酸酯、丙二醇和邻苯二甲酸二丁酯。特别是,阴离子羧基丙烯酸聚合物通常含有10-25%重量比的塑化剂,尤其是邻苯二甲酸二丁酯、聚乙二醇、柠檬酸三乙酯和三乙酸甘油酯。应用常规的包膜技术例如流体床或沃斯特包膜机或喷雾或锅包衣来涂覆膜衣。膜衣的厚度必须足以确保口服剂型维持完整,直到到达肠道中所欲的局部递送处。
[0116]
除了塑化剂外可于膜衣中加入色剂、表面活性剂、抗黏附剂、消泡剂、润滑剂(例如巴西棕榈蜡或peg)及其他的添加剂,用以增加溶解或分散膜衣物质,并提高膜衣效能及该包衣产品。
[0117]
为了加速肠溶膜衣溶解,可涂覆一半厚度的双层肠溶膜衣聚合物(例如,eudragit l30 d-55),且内层肠衣在10%柠檬酸的存在下可具有至高ph 6.0的缓冲剂,接着最后的标准eudragit l 30 d-55层。相较于涂覆以无缓冲作为单一层的类似的涂膜系统,涂覆二层肠溶膜衣,各一半厚度的典型肠溶膜衣,liu和basit能加速肠衣溶解(liu,f.and basit,a.journal of controlled release.147(2010)242-245.)。
[0118]
肠溶膜衣的完整性可,例如通过药物在微颗粒中的降解来测量。包覆肠溶膜衣的剂型或药丸可如usp中所述以溶解试验先在胃液中及分开地于肠液中检测,测定其功能。
[0119]
含有公开化合物的包覆肠溶膜衣的片剂和胶囊制剂可通过本领域中熟知的方法来制造。例如,含有文中所公开化合物的片剂可以含有邻苯二甲酸二乙酯、异丙醇、滑石和水的膜溶膜衣溶液使用侧面通气包衣锅(freund hi-coater)来包覆肠溶膜衣。
[0120]
或者,可如下制备可并入片剂中或并入胶囊中的包括包覆肠溶膜衣药丸的多单位
剂型。
[0121]
核心物质:用于个别肠溶膜衣层覆药丸的核心物质可根据不同原理建构。以层覆活性剂的核心粒子(亦即,btk抑制剂化合物和/或其药学上可接受的盐),任选地混合碱性物质或缓冲剂,可用作为核心物质供进一步加工。层覆上活性剂的核心粒子可包括单独或混合的不同氧化物、纤维素、有机聚合物和其他物质的不溶于水的核心粒子,或包括单独或混合的不同无机盐类、糖、糖丸(non-pareils)和其他物质的水溶性核心粒子。再者,所述核心粒子可包括晶体、团块、压缩物等形式的活性剂。核心粒子的大小就本文并非必要,但可在大约0.1至2mm间变化。层覆上活性剂的核心粒子为通过粉末或溶液/悬浮液层覆使用例如造粒或喷雾包膜层覆设备来制造。
[0122]
在核心粒子层覆之前,活性剂可与另外的组份混合。这些组份可为单独或混合的粘结剂、表面活性剂、填充剂、崩解剂、碱性添加剂或其他和/或药学上可接受成份。粘结剂为例如聚合物,例如羟丙基甲基纤维素(hpmc)、羟丙基-纤维素(hpc)、羧甲基纤维素钠、聚乙烯吡咯烷酮(pvp)或糖、淀粉或其他具有黏性的药学上可接受物质。适合的表面活性剂可参见药学上可接受非离子或离子表面活性剂的群,例如,举例而言月桂基硫酸钠。
[0123]
或者,任选地与适合组成份混合的活性剂可调配成核心物质。该核心物质可通过挤压/滚圆、球状化或压制利用常规的加工设备来制造。经调配的核心物质的大小大约为介于0.1至4mm之间,及例如介于0.1至2mm之间。制造的核心物质可进步层覆上包括活性剂的另外的成份和/或用于进一步加工。
[0124]
活性剂与药物成份混合以得到优选的处理和加工性质及最终制剂中适合的活性剂浓度。可使用药物成份,例如填充剂、粘结剂、润滑剂、崩解剂、表面活性剂和其他药学上可接受添加剂。
[0125]
或者,前述核心物质可通过使用喷雾干燥或喷雾冷凝技术来制备。
[0126]
肠溶膜衣层:在个别药丸形式的核心物质上涂覆肠溶膜衣层之前,该药丸可任选地覆盖一或多层包括药物赋形剂,任选地包括碱性化合物例如ph-缓冲化合物的分离层。这些分离层,为分开核心物质与外层肠溶膜衣。这些保护活性剂的核心物质的分离层应为水溶性或在水中快速崩解。
[0127]
可通过包膜或分层制程以适合的设备,例如包衣锅、包膜造粒剂或流化床装置使用水和/或有机溶剂进行包膜程序,任选地于核心物质上涂覆分离层。或者,可通过使用粉末包膜技术于核心物质上涂覆分离层。用于分离层的物质为药学上可接受化合物,例如,举例而言,单独或混合的糖、聚乙二醇、聚乙烯吡咯烷酮、聚乙烯醇、聚乙酸乙酰酯、羟丙基纤维素、甲基纤维素、乙基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠、肠衣聚合物的水溶性盐类和其他。亦可在分离层中包括添加剂,例如塑化剂、色剂、颜料、填充剂、防黏剂和抗静电剂,例如,举例而言,硬脂酸镁、二氧化钛、滑石和其他添加剂。
[0128]
当核心物质涂覆任选的分离层时,其可建构各种厚度。分离层的最大厚度通常仅受限于加工条件。这些分离层可作为扩散屏障及作为ph-缓冲区。任选地涂覆的分离层就本文的实施方案并非必需的。然而,分离层可提升活性物质的化学稳定性和/或新的多单位片剂剂型的物理性质。
[0129]
或者,分离层可通过涂覆在核心物质上的肠溶膜衣聚合物和核心物质上的碱性反应化合物间的反应,原位形成。因此,所形成的分离层包括位于形成盐的位置上在肠溶膜衣
聚合物和碱性反应化合物间形成的水溶性盐。
[0130]
通过使用适合的包衣技术可将一或多层膜衣涂覆在核心物质上或覆盖分离层的核心物质上。肠溶膜衣层物质可分散或溶于水或适合的有机溶剂中。可分开或组合使用下列一种或多种的肠衣层聚合物,例如甲基丙烯酸共聚物、纤维素乙酸邻苯二甲酸酯、羟丙基甲基纤维素邻苯二甲酸酯、羟丙基甲基纤维素乙酸琥珀酸酯、聚乙烯乙酸邻苯二甲酸酯、纤维素乙酸偏苯三酸酯、羧甲基乙基纤维素、虫胶或其他适合的肠溶膜衣聚合物的溶液或分散液。
[0131]
肠溶膜衣层含有药学上可接受塑化剂用以得到所欲的机械性质,例如肠溶膜衣层的弹性和硬度。这些塑化剂有,例如,但不限于三乙酸甘油酯、柠檬酸酯、邻苯二甲酸酯、癸二酸二丁酯、鲸蜡醇、聚乙二醇、聚山梨醇酯或其他塑化剂。
[0132]
塑化剂的量就有关选择的肠溶膜衣层聚合物、选择的塑化剂和该聚合物的涂覆量,针对各肠衣层配方进行优化,以此方式调整机械性,亦即肠衣层的柔软性和硬度,例如以维氏硬度(vickers hardness)为例,使得一所欲的片剂在丸粒压制成片剂期间,该覆盖肠衣层的丸粒的抗酸性不会显著降低。塑化剂的量通常为肠溶膜衣层聚合物的5%重量比以上,例如15至50%及另外20-50%。添加剂,例如分散剂、色剂、颜料聚合物,例如聚(乙基丙烯酸酯,甲基丙烯酸甲酯)、防黏剂和消泡剂一可包括在肠溶膜衣层内。可加入其他的化合物用以增加膜厚度及降低酸性胃酸扩散至易感染酸的物质中。涂覆的肠衣的最大厚度通常仅受限于加工条件和所欲的溶解样貌。
[0133]
外套层(over-coating layer):覆盖上肠衣层的丸粒可任选地进一步覆盖一或多层之外套层。该外套层应为水溶性或快速崩解于水中。外套层可通过涂膜或层覆程序以适合的设备例如包衣锅、涂膜造粒机或流化床装置,使用水和/或用于涂膜或层覆处理的有机溶剂,涂覆于层覆肠溶膜衣层的丸粒上。用于外套层的物质为由药学上可接受的化合物中选出,例如单独或混合的糖、聚乙二醇、聚乙烯吡咯烷酮、聚乙烯醇、聚乙酸乙烯酯、羟丙基纤维素、甲基纤维素、乙基纤维素、羟丙基甲基纤维素、羧甲基纤维素钠和其他。添加剂,例如塑化剂、色剂、颜料、填充剂、防黏剂和抗静电剂,例如硬脂酸镁、二氧化硅、滑石和其他添加剂亦可包括在外套层中。外套层可防止层覆肠溶膜衣的丸粒可能的凝结,再者其可保护肠溶膜衣层在压制处理期间免于碎裂并增进成片处理。涂覆之外套层的最大厚度通常仅受限于加工条件和所欲的溶解样貌。外套层亦可用作为片剂的膜溶膜衣层。
[0134]
软式明胶胶囊的肠溶膜衣可含有乳液、油、微乳液、自乳化系统、脂质、三甘油酯、聚乙二醇、表面活性剂、其他增溶剂及诸如此类和其组合,用于溶化活性剂。软式明胶胶囊的柔软性通过残余水和塑化剂维持。此外,对于明胶胶囊,明胶可溶于水因此喷雾必须以相当低的相对湿度比率来进行,例如可以流化床或沃斯特法来进行。此外,干燥应在不会移除残余水或塑化剂而造成胶囊外壳碎裂下进行。就软式明胶胶囊的肠溶膜衣优化的市售混拌物,例如instamodel epd(肠溶膜衣聚合物分散液)可得自ideal cures,pvt.ltd.(mumbai,india)。对于一实验室规模,肠衣胶囊可通过:a)于一烧瓶中旋转胶囊或将胶囊浸入以最低可能温度缓和加热含有塑化剂的肠衣物质或b)用一实验室规模的喷雾剂/流化床及然后干燥来制备。
[0135]
对于水性活性剂,将药物并入乳液的水相中可能是特别有益的。此“油包水”乳液提供适合该药物的生物物理环境并可提供一油-水接口,其可保护药物免于可能降解药物
的ph或酶的有害效应。另外,此油包水制剂可提供一脂质层,其可有利地与身体细胞中的脂质相互作用,且可增加制剂分溶进入细胞膜。此分溶可增加此调配中药物吸附入血液循环且因此可增加药物的生物利用度。
[0136]
在某些实施方案中,油包水乳液含有一由媒剂或长链羧酸或其酯类或醇类、表面活性剂所组成的油相,及一主要含有水和活性剂的水相。
[0137]
媒剂和长链羧酸为范围从c8至c
22
具有至高三个不饱和键(以及支链)的媒剂和长链羧酸。饱和直链酸类的实例有正十二烷酸、正十四烷酸、正十六烷酸、己酸、辛酸、癸酸、月桂酸、肉豆蔻酸、棕榈酸、硬脂酸、花生酸、山嵛酸、二十八酸和蜜蜡酸。不饱和单烯烃直链单羧酸亦是有用的。这些酸的实例有油酸、鳕油酸和芥酸。不饱和(多烯烃)直链单羧酸亦是有用的。这些酸的实例有亚麻油酸、蓖麻油酸、次亚麻油酸、花生四烯酸和萝炔酸。有用的支链酸类包括,例如,二乙酰酒石酸酯。不饱和烯烃链亦可经羟基化或乙氧基化用以防止氧化或改变表面性质。
[0138]
长链羧酸酯的实例包括,但不限于所述来自下列的群:单硬脂酸甘油酯;单棕榈酸甘油酯;单硬脂酸甘油酯和单棕榈酸甘油酯的混合物;单亚麻油酸甘油酯;单油酸甘油酯;单棕榈酸甘油酯、单硬脂酸甘油酯的混合物,单油酸甘油酯和单亚麻油酸甘油酯的混合物;单次亚麻油酸甘油酯;单鳕油酸甘油酯;单棕榈酸甘油酯、单硬脂酸甘油酯、单油酸甘油酯、单亚麻油酸甘油酯、单次亚麻油酸甘油酯和单鳕油酸甘油酯的混合物;乙酰甘油酯,例如蒸馏的乙酰单甘油酯;丙二醇单酯、蒸馏的单甘油酯、硬脂酰乳酰乳酸钠和二氧化硅的混合物;d-α生育酚聚乙二醇1000琥珀酸酯;单-和二-甘油酯的混合物,例如atmul;硬脂酰乳酰乳酸钙;乙氧基单-和二-甘油酯;乳酸单-和二-甘油酯;甘油和丙二醇的乳酰羧酸酯;长链羧酸的乳酸酯;长链羧酸的多甘油酯、长链羧酸的单-和二-丙二醇酯;硬脂酰乳酰乳酸钠;单硬脂酸山梨酸酐酯;单油酸山梨酸酐酯;长链羧酸的山梨醇酐酯;琥珀酸单甘油酯;硬脂酰基柠檬酸单甘油酯;硬脂酰庚酸酯;鲸蜡酯;硬脂酰辛酸酯;c
8-c
30
胆固醇/羊毛甾醇酯;及蔗糖长链羧酸酯。自乳化长链羧酸酯的实例包括来自下列群的酯类:硬脂酸酯、棕榈酸酯、蓖麻油酸酯、油酸酯、山嵛酸酯、肉豆蔻酸酯、月桂酸酯、辛酸酯和己酸酯。在某些实施方案中,油相可包括2或更多的长链羧酸或其酯类或醇类的组合。在某些实施方案中,可使用中链表面活性剂且该油相可包括辛酸/癸酸三甘油酯和辛酸的c8/c
10
单-/二-甘油酯、辛酸甘油酯或丙二醇单辛酸酯或其混合物。
[0139]
可使用的醇类举例而言有上述作为例示的羧酸的羟基形式以及硬脂醇。
[0140]
表面活性剂是可累积在亲水性/疏水性(水/油)表面并在界面降低表面张力的长链分子。因此,其可使乳液稳定。在某些实施方案中,表面活性剂可包括:(聚氧化乙烯山梨醇酯)家族的表面活性剂,(山梨醇酐长链羧酸酯)家族的表面活性剂,(环氧乙烷或环氧丙烷嵌段共聚物)家族的表面活性剂,和(均为聚乙二醇甘油酯)家族的表面活性剂,油酸、硬脂酸、月桂酸和其他长链羧酸的山梨醇酐酯、泊洛沙姆(聚乙二醇-聚丙二醇嵌段共聚物或),其他山梨醇酐或蔗糖长链羧酸酯,单和二甘油酯、辛酸/癸酸三甘油酯的peg衍生物及其混合物,或二或更多上述的混合物。在某些实施方案中表面活性剂相可包括聚氧乙烯(20)山梨醇酐单油酸
酯(tween)和山梨醇酐单油酸酯(span)。
[0141]
水相可任选地包括悬浮在水中和缓冲液中的活性剂。
[0142]
在某些实施方案中,这些乳液为粗乳液、微乳液和液体晶体乳液。在其他的实施方案中,这些乳液可任选地包括渗透增强剂。在其他的实施方案中,可使用喷雾干燥悬浮液或含有微粒子或纳米粒子的微胶化微乳液、粗乳液或液体晶体。
[0143]
在某些实施方案中,文中所述的固体剂型为无肠溶膜衣时间推迟释放的剂型。术语“无肠溶膜衣时间推迟释放”如文中所用是指递送使得释放药物可在肠道中某些一般可预测的位置进行,比在无延迟释放选项下进行更长远。在某些实施方案中延迟释放的方法为在一段设定的时间后,变得可穿透、溶解、破裂和/或不再完整的膜衣。在时间延迟释放剂型中的膜衣可具有一固定时间溶蚀,之后释放药物(适合的膜衣包括聚合物膜衣,例如hpmc、peo及诸如此类),或具有一由超级崩解剂或渗透剂或水引诱剂所组成的核心,例如盐、亲水性聚合物,典型地聚环氧乙烷或烷基纤维素,盐类例如氯化钠、氯化镁、乙酸钠、柠檬酸钠,糖例如葡萄糖、乳糖或蔗糖或诸如此类,其经由半透膜及水,或产气剂例如柠檬酸和有或无例如柠檬酸或任何前述酸类的酸的碳酸氢钠,并入剂型中。该半透膜,大多是药物及渗透剂不可穿透的,是水可穿透的,水以近乎固定的速率穿透进入剂型中,以增加压力并在一段所欲的延迟时间内膨胀压超过一特定阈值后碎裂。药物通过此膜的渗透力应低于水的1/10且在一实施方案中为低于1/100水渗透力。或者,膜可通过浸出水性可浸出物一段所欲的延迟时间变成多孔性的。
[0144]
渗透剂型已描述于theeuwes美国专利第3,760,984且渗透破裂剂型描述于bake美国专利第3,952,741号中。此渗透破裂剂型可提供单脉冲释放或使用不同时序的不同装置则提供多脉冲。渗透破裂的时序可通过选择聚合物和围绕在含有药物和渗透剂或引诱剂的核心的半渗透膜的厚度或面积来控制。因为剂型中的压力随着加入的渗透水增加,所以膜扩大直到破裂点,及然后药物释放。或者,可通过膜中具有较薄、较弱区域,或通过加入弱化物质于涂覆膜的区域,在膜中制造特定的破裂区域。某些可用作为半渗透膜、具有高度水渗透性的优选聚合物为纤维素乙酸酯、纤维素乙酸丁酸酯、纤维素硝酸酯、交联聚乙烯醇、聚氨酯、尼龙6、尼龙6.6和芳香类尼龙。纤维素乙酸酯是特别优选的聚合物。
[0145]
在另外的实施方案中,在肠衣至少部分溶解后才开始延迟其释放时间的延迟膜衣是由与水接触才开始随时间逐渐溶蚀的亲水性、可溶蚀聚合物所组成。这些聚合物的实施例包括纤维素聚合物及其衍生物,包括(但不限于)羟烷基纤维素、羟甲基纤维素、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、羧甲基纤维素、微晶纤维素;多糖类及其衍生物;聚环氧乙烷,例如聚环氧乙烷或聚乙二醇,尤其是高分子量聚乙二醇;几丁聚糖;聚(乙烯醇);黄原胶;马来酸酐共聚物;聚(乙烯吡咯烷酮);淀粉及以淀粉为基底的聚合物;麦芽糊晶;聚(2-乙基-2-噁唑啉);聚(乙二胺);聚氨酯;水凝胶;交联聚丙烯酸;及任何前述的组合或混合物。
[0146]
某些适合形成可溶蚀膜衣的优选的可溶蚀亲水性聚合物为聚(环氧乙烷)、羟丙基甲基纤维素及聚(环氧乙烷)和羟丙基甲基纤维素的组合物。聚(环氧乙烷)在文中使用是指未经取代的环氧乙烷直链聚合物。聚(环氧乙烷)聚合物的分子量范围可从约105道尔顿至约107道尔顿。聚(环氧乙烷)聚合物的优选的分子量范围为从约2x105至2x106道尔顿且在市面上可从dow chemical公司(midland,mich.)购得,称为sentryr polyox
tm
水溶性树脂,nf
(national formulary)等级。当使用较高分子量的聚(环氧乙烷)聚合物的分子量范围时,亦可包括促进此膜衣溶蚀或崩解的其他亲水试剂,例如盐类或糖类,如葡萄糖、蔗糖或乳糖。
[0147]
时间延迟剂型可为机械性药片,例如胶囊或ph敏感性胶囊,可在一预编程的时间之后或当其接收到一可传送的信号时或是一但离开胃后,释放该药物。
[0148]
制剂中本文化合物的量可在本领域技术人员所用的全范围内变化。典型地,以重量百分比为基准,此制剂将含有从约0.01至99.99重量%的btk抑制剂化合物(以总制剂为基准),其余的为一种或多种的适合药物赋形剂。在一实施方案中,化合物以约1至80重量%的量存在。
[0149]
就清楚和理解的目的,上述公开内容已经通过说明和举例的方式进行了一些详细描述。因此,应理解上述说明书希望为说明性的且并非限制性。本文的范围因此不应仅就有关上述说明来判定,而应参考后续所附的权利要求书以及所述权利要求书应享有的全部均等范围来判定。实施例
[0150]
提供下列实施例用以说明特定公开的实施方案且不应理解为在任何方面限制本文的范围。在下列所公开的实施例中,如上所定义的btk抑制剂亦可互换称为“该化合物”或“该药物”。实施例1-复发型多发性硬化症中btk抑制剂的剂量探索研究实施例1.1-导论和概论
[0151]
此第2b期研究的目的是用于定义安全、最佳的btk抑制剂的剂量。btk抑制剂的提议作用机制,如mri所测,为抑制ms中新的活动性脑病灶形成且因此预计在ms病患中以进一步的试验验证临床效用。此研究为通过测量与发炎有关的钆(gd)-增强的t1-高信号病灶数目的变化,评估剂量反应。已建立此放射线照相结果作为ms枢纽试验中临床效用的高可靠性预测生物标记并已验证为第3期登录研究中临床效用(降低arr)的预测生物标记(sormani et al,ann neurol.2009;65(3):268-75;sormani et al,neurology,2010;75(4):302-9))。以4剂量层级和一短期安慰剂为基准,以2-步骤统计法,评估病灶抑制的剂量反应。通过评估mri所测量的抑制新的活动性脑病灶形成,评估btk抑制剂相对于安慰剂的效用。此研究亦找出btk抑制剂在患有rms的参与者中的安全性和耐受的特性。
[0152]
本研究为利用许多的次要结果测量致力于收集btk抑制剂在神经发炎上潜在利益的额外数据。
[0153]
预期以探索性评估,例如分析血清中nfl量和先进造影法,开始建立btk抑制剂活性对神经发炎和神经退化的证据,以及对于髓鞘再生和组织保存的潜在效用。图1显示整体研究设计图而表1显示活动时程(soa)。
[0154]
治疗目标和终点为如表2所示。表2:目标及终点
[0155]
测量的适当性
[0156]
脑中发炎活动性的核磁共振成像(mri)标记以如大多数的rms临床试验的方式收集。使用新的gd-增强的t1-高信号病灶数作为主要终点,用于评估btk抑制剂的效用。因为ms造成血脑屏障漏洞,因此gd造影剂堆积在脑组织中与ms病患的发炎活动性有关。已建立此放射线照相结果作为ms的枢纽研究中临床效用的高可靠性预测生物标记。使用集中审查来鉴定之前的mri未出现新的gd-增强的t1-高信号病灶。亦使用gd-增强的t1-高信号病灶总数作为次要终点,用来检测任何对先前存在的发炎病灶的效用。亦以集中审查评估新的和扩大的t2病灶数(一种rms中发炎活动性和脑组织破坏的标记),用以收集与btk抑制剂的效用有关的另外数据。亦评估t2病兆的总体积(ms负荷)和t1-减弱信号病灶数(黑洞)作为与效用有关的支持性数据。
[0157]
核磁共振成像(mri)测量包括脑容量的变化,其被视为cns退化的标记,但亦与rms病患中的发炎事件相关。数种ms药物以其减缓脑萎缩的能力为人所知,其以找寻可能的信号来评估。
[0158]
临床复发是rms主要的临床表现。复发相关的终点(arr,无复发参与者的比例)广泛地用作为临床试验的终点。虽然此试验的持续时间短无法期待发生复发的给剂组间的显著差异且在ppms中复发被认为不常发生,但由于其临床的重要性及希望收集额外的功效数据而进行评估。
[0159]
此edss在临床试验及例行设定中被广泛用于测量神经失能(kurtzke jf,
neurology.1983;33(11):1444-52)。在此研究期间内预计并无大的变化,但用作为功效的支持性数据。实施例1.2-研究设计
[0160]
整体设计:2b期、随机双盲、安慰剂对照、交叉的剂量范围研究,用以研究mri效用和12周btk抑制剂给药的的安全性。经诊断患有rms者可以登记,只要其符合所有的纳入标准且无排除标准。
[0161]
交叉之前使用互动语音/网络回复系统(ivrs/iwrs),所有的参与者集中分配成8臂中的其中之一(2个群组的各4个剂量组相同比例,开始btk抑制剂(第1群组)或安慰剂(第2群组)期。
●
在各群组内,参与者通常以遮盲方式,随机平均分配于4个btk抑制剂剂量,每日一次5、15、30或60mg的其中之一。
●
第1群组:参与者接受其中一种btk抑制剂剂量先历时12周,然后转换至安慰剂历时4周。
●
第2群组:参与者先接受安慰剂历时4周,然后转换至其中一种btk抑制剂剂量历时12周。
[0162]
在完成双盲治疗期后,参与者可选择进入长期安全性(lts)追踪研究,用以评估btk抑制剂的安全性和耐受性。
[0163]
参与者人数:大约160位经筛选至随机分配大约120位参与者(以25%筛选失败率为基础)而进行研究介入有大约105位可评估的参与者(以大约15%中辍率为基础,提供btk抑制剂的各剂量组至少26位参与者)完成12周的btk抑制剂治疗。来自第2群组的参与者(n=60),在转换至btk抑制剂之前为接受4周的安慰剂,提供可用于估算剂量反应曲线和与安慰剂相比较的数据。此方法以在安慰剂下于12周期间理论上固定比率的新的gd-增强的t1-高信号病灶的假设为基础。此方法将研究参与者的安慰剂暴露减至最低。处理安慰剂数据和分析以及另外的详情包括样本大小的决定的简要说明,提供于实施例1.14中。
[0164]
介入组和持续时间:在12-周的btk抑制剂治疗之后或之前(分别为第1和第2群组)导入4-周的安慰剂。参与者以等比例随机分配至各8组中(各2个群组中的4个剂量组)。研究介入的概述请参见表5。
[0165]
基本原理:本研究对于剂量和给药顺序是遮盲的。其关注剂量探索但亦考虑将参与者对安慰剂的暴露降至最小的需求。因此,剂量范围使用4种剂量来评估:每日一次5、15、30和60mg。此外,为了最小化对安慰剂的暴露同时维持遮盲研究者和参与者,将各参与者分派于发生在研究的最前面或最后面4周期间的4周安慰剂期。在12周的btk抑制剂治疗之后或之前(分别为第1和第2群组)导入4-周的安慰剂期。将参与者随机分配成8臂中的其中之一(各2个群组等比例4个剂量组。安慰剂的给药时间限制为4周用以最小化安慰剂暴露;交叉的设计使得所有的参与者能以btk抑制剂治疗。此交叉设计对给药介入为遮盲的并在研究开始时允许更客观的安全事件及效用终点的评估。12周的btk抑制剂治疗持续时间应能检测其对抑制新的gd-增强的t1病灶形成的效应。在一rms病患的依伏替尼(evobrutinib)研究的最新近通讯中确认了12周已可观察到有意义降低这些病灶(merck press release-merck kgaa,darmstadt,germany,announces positive phase iib results for evobrutinib in relapsing multiple sclerosis.7 mar 2018.)。
[0166]
剂量疗法:本研究选择的剂量范围由数种评估来反映。首先,在临床前动物(小鼠、大鼠和狗)中意欲用来翻译btk抑制剂的btk占有率的异速生长模型预测出人类介于1至100mg每日一次的最佳剂量范围。第二,在人类外围血液单核细胞(pbmc)的第1期的btk占有率的多递增剂量测量,显示每日一剂7.5mg的btk抑制剂受体渐趋近饱和,其中较高剂量更快接近饱和。最后,绝对cd19 b-细胞计数的测量显示相对于基线高达80%的剂量依赖增加(在第4天观察到最大值)。从文献中预测btk-引发的循环中b细胞增加,因为btk抑制改变细胞表面黏附分子表达,导致从淋巴结退出(burger ja et al.,nat rev cancer.2018;18(3):148-67)。就此效应的剂量反应关系在每日一次约30mg时为最大。考虑所有这些因素,设定介于5至60mg每日一次的剂量范围用来提供捕捉rms中btk抑制剂的最适剂量的最佳机会。
[0167]
研究结束定义:若参与者已完成所有的研究期包括最后返诊,则该参与者视为已完成本研究。研究结束定义为本研究的最后一位参与者完成最后返诊的日期。实施例1.3-研究族群实施例1.3a-纳入标准
[0168]
仅在适用所有下列如表3中所示的标准时,参加者才可被纳入本研究。表3-纳入标准实施例1.3b-排除标准
[0169]
若适用任何下列如表4中所示的标准,则将参加者从本研究排除。
表4-排除标准
实施例1.4-研究介入
[0170]
研究介入定义为根据研究方案打算施用研究参与者的任何研究性介入、市售产品、安慰剂或医疗设施。实施例1.4-施用的研究介入(study intervention)
[0171]
本研究介入包括imp和非研究用药品(nimp)。为了保持遮盲,参与者以受药不明的方式每天一次接受4片的btk抑制剂和/或安慰剂。介入的详情提供于表5中。表5-施用的研究介入的概述
[0172]
nimp:使用放射学、信号强化、静脉内(iv)显影剂,进行t1对比强化mri系列。使用当地批准的显影剂。实施例1.4a1-最小化偏差的方法:随机分配及遮盲
[0173]
在转换前,使用ivrs/iwrs,所有的参与者集中分配成8臂中的其中之一(2个群组各自4个剂量组相同比例,开始btk抑制剂(第1群组)或安慰剂(第2群组)期。在本研究中参与者不能随机分配超过一次。在研究开始前,于各处提供ivrs的电话号码和招集指示和/或iwrs的登录信息和指示。研究介入在概述于活动时程表(表1)的研究返诊时分配。归还的研
究介入不应再分配给参与者。
[0174]
破盲(ivrs/iwrs):ivrs/iwrs与破盲指令编定程序。若紧急时,研究者全权负责决定是否必要解盲参与者治疗的分派。参与者的安全必须是做此决定的首要考虑。若研究者决定必须解盲,则研究者应在解盲参与者的治疗分派前,尽一切努力联络赞助者,除非可能延迟该参与者的紧急治疗。若参与者的治疗分派解盲了,则必须在破盲后24小时内通知赞助者。破盲的日期和理由必须记录在原始数据文件中及案例报告表,任选而定。
[0175]
本研究对于剂量和btk抑制剂-安慰剂给药顺序遮盲。不同剂量的btk抑制剂片剂和安慰剂是相同的。由于道德上的考虑,安慰剂持续时间限制在4周,此举能在研究期开始时更客观评估安全性事件,及加入临床疗效评估的客观性。
[0176]
研究者无法取得mri数据,除非有传达任何非-ms-相关的发现,用以评估参与者的安全。各研究处的放射线服务负责将任何mri上的非-ms发现定期向研究者提出报告。
[0177]
使用独立数据监查委员会(idmc)定期地监查本研究的安全性。由一解盲独立统计员提供解盲数据供idmc审查。研究团队成员、研究者和研究参与者无法取得解盲数据。实施例1.4b-并用治疗
[0178]
参与者在招募时接受的或在研究期间接受的任何药品和疫苗(包括非处方或处方药、维生素和/或草药补充品)与使用理由、施用日期(包括开始和结束日期)以及给剂信息(包括剂量和频率)一起记录。
[0179]
就于招募前4周期间接受的所有先前医疗,以及所有先前ms治疗和被认为在评估ms或并发疾病是临床上重要的治疗,收集相同资料。高剂量的糖皮质类固醇的ms复发的标准治疗是允许的。此类治疗依循当地规则。
[0180]
除了排除表4中的药物外,在整个研究中禁止下列医疗:-其他的ms疾病改善治疗-乙酰水杨酸(阿司匹林)-抗血小板药物(例如克洛平格(clopidogrel))-抗凝血剂,包括:华法林、肝素,包括低分子量肝素(抗凝血酶)、达比加群、阿哌沙班、依度沙班、利伐沙班(rivaroxaban)。
[0181]
就乙酰胺酚(paracetamol)/乙酰胺酚(acetaminophen),在研究期间的任何时间,≤3克/天的剂量是允许的。若就现存的医疗状况或新的事件临床上需要治疗,在研究期间可给予短期(至高5天)的建议剂量的nsaid(乙酰水杨酸以外)。研究者于crf中记录nsaid的使用(及任何其他并用药物)。
[0182]
体外实验和计算机仿真(in silico)模型已验证降胃酸剂可能降低btk抑制剂的血浆暴露。应避免使用质子泵抑制剂(例如,奥美拉唑(omeprazole))。使用抗酸剂(例如,碳酸钙)应与btk抑制剂给剂错开,抗酸剂给药应在btk抑制剂给剂之前2小时以上或2小时之后进行。使用h2-受体拮抗剂(例如,雷尼替汀(ranitidine))亦应与btk抑制剂给剂错开,h2-受体拮抗剂应在btk抑制剂给剂之前10小时以上或2小时之后进行。可能经由降低胃酸影响btk抑制剂的血浆暴露的实施例药物列表请参见表13。
[0183]
以临床前的药物代谢研究为基础,btk抑制剂为cyp3a和cyp2c8同功酶的基质,且因此,若与其他引发或抑制cyp3a和/或cyp2c8代谢的药物共同给药可能改变btk抑制剂的血浆暴露。此项到目前尚未在人类中研究过,且因此尽可能应避免强力抑制或引发cyp3a或
cyp2c8的药物。不能使用的药物列表请参见表12。实施例1.4c-剂量调整
[0184]
在本研究中并未预想降低剂量。参与者、研究者和赞助者团队皆不知分发的剂量。若由于ae而实际上有需要,则可能必须打断或永久中断治疗(实施例1.4e和1.8)。实施例1.4d-研究结束的介入
[0185]
对完成本研究第16周返诊的参与者提供一隔开、开放的lts研究。在完成双盲治疗期后,参与者皆已招募入dri研究且所有后续的参与者给予进入一lts追踪研究的选项,用以评估btk抑制剂的安全性和耐受性。实施例1.4e-中断研究介入和参与者中断/退出
[0186]
同意退出治疗应与(另外)同意退出追踪返诊和同意退出的不参加者接触(例如,医疗记录查询)追踪做区分。各研究处应记录任何同意退出的案例。实施例1.4e1-中断研究介入
[0187]
确定中断:只要可能应持续imp。若停止imp,应确定该停止是否是暂时性的;确定中断应为最后手段。任何imp中断完整记录在ecrf中。在任何情况下,参与者应留在本研究中越久越好。确定的介入中断是研究者不会让参与者在研究期间的任何时间再暴露于imp,或基于任何理由参与者不再暴露于imp的确定决定有关的任何介入中断。当参与者符合10.6节所述的任一条件,或研究者相信基于参与者的最佳利益时,因肝功能异常而中断研究介入应由研究者考虑。若在纳入后于ecg中鉴定出临床上显著发现(包括,但不限于使用弗里德里西亚公式(fridericia’s formula)[qtcf]校正的qt间距中与基线相比的变化),则研究者或合格的指定人员决定该参与者是否可以继续本研究或需要就参与者管理做任何改变。心脏专科医师所做的ecg状况审查需要列入作为因ecg变化而决定明确中断研究介入的考虑。记录在收集时间所打印的ecg审查。任何新的临床上相关发现报告为一ae。
[0188]
就在介入中断时(治疗结束的返诊)所收集的数据和追踪及任何需要完成的进一步评估,请参见soa(表1)。在该有关的参与者做出决定确定中断介入前,任何异常的实验室数值或ecg参数请在24小时后立即再检查进行确认。若永久中断介入,则进行结束治疗的返诊。
[0189]
根据此方案中所指定的研究程序追踪参与者直到研究完成,或如本方案所指定,任何所追踪的ae恢复或稳定,以时间后到者为准。若可能,及在确定中断介入后,使用正常计划进行imp的最后治疗日,包括pk样本,评估参与者。详情提供于soa(表1)中。当认为确定时,由研究者于ecrf的适当页面,记录所有确定中断介入的案例。
[0190]
因为疑似ae和/或实验室数据异常和/或ecg异常,可被研究者视为暂时的中断介入。就所有暂时的介入中断,应由研究者将中断期间记录在ecrf的适当页面。由研究者决定的暂时介入中断相当于有>1个剂量未施用参与者。
[0191]
一旦研究者根据其最佳医疗判断认为有关事件的发生不太可能是imp的责任且若仍然符合选择的研究标准,则于紧密和适当的临床和/或实验室监测下进行再启动imp介入(参照表3)。实施例1.4e2-参与者中断/退出
[0192]
参与者可能依其本身要求在任何时间退出,或可能因安全性、行为、遵从性或给药理由,在研究者的自由裁量权下在任何时间退出。
multiple sclerosis functional composite measure(msfc):an integrated approach to ms clinical outcome assessment,”national ms society clinical outcomes assessment task force.mult scler.1999;5(4):244-50)。
[0202]
在每次的返诊时依据soa(表1),将病灶数与先前mri扫描的病灶数相比较,评估新的t1 gd-增强的高信号以及新的和扩大的t2病灶。除非另有指出,否则使用基线脑mri作为评估所有mri-衍生的指标评估点的参照。基线mri是在随机分配诊察前最后进行的mri。由集中审查脑mri扫描,确保标准化指针评估点的评估。对所有的mri-衍生的指标评估进行遮盲的集中审查。核磁共振成像审查员对于治疗分派和其他参与者的数据并不知情。若研究者怀疑有脊椎ms病灶,则可能需要脊椎mri。于当地评估脊椎mri并报告于ecrf中。脊椎mri不用进行集中审查。
[0203]
探索性效用评估的核磁共振成像为使用区域或整个脑容积评估,另外的t1和t2造影分析,以及例如磁转移比率和敏感性加权成像系列。实施例1.6b-多发性硬化症复发
[0204]
疑似多发性硬化复发的非时程内的评估返诊:参与者被嘱咐向研究者立即报告新的神经征候和再发生先前征候或先前征候恶化。收集任何报告的征候。若参与者报告一可能与复发相符的征候,则尽早安排一与研究者的非时程内的评估返诊(尽可能在征候发生的7天内)。研究者评估所报告的情节是否与ms复发的定义相符(参见实施例1.6b)。若与ms复发的定义相符或有任何质疑且无法排除复发,则应进行一edss评估。非时程内的返诊活动详述于soa(表1)中,若是由ms以外的病理所致,则需要调整,且需要额外的检查或实验室检验供安全性追踪和最佳的治疗选择。
[0205]
多发性硬化症复发:就本研究的目的,ms复发定义为神经学检查上真实改变的急性、新的神经征候或先前的神经征候恶化:
●
归因于ms
●
持续≥24小时,及
●
在正常体温时出现(亦即,无感染、过度运动或过高的周围温度)
[0206]
注意:征候或征象的恶化或在发生,可能合理地归因于由于药物在先前脱髓鞘路径中暂时性的传导障碍(例如注射干扰素β后数小时鲜少发生),核心体温升高(乌托夫现象(uhthoffphenomenon)),或全身性细胞因子释放(例如阿仑单抗给药时发生)不能视为复发。实施例1.6c-扩展失能状况量表评估
[0207]
研究者如soa(表1)中所示进行edss(kurtzke jf,neurology.1983;33(11):1444-52)。
[0208]
研究者在标准神经检查的背景下评量功能系统并将这些评比依照edss报告指南与参与者的活动性、步态和使用的辅具信息一起报告。在各7个功能区(视觉、脑干、锥体[运动]、小脑[协调]、感觉、大脑和肠/膀胱)进行标准的神经学征候的edss评估。亦对移动进行评分作为部分的评估。可任选地做疲劳的评估,但不能作为edss的得分。实施例1.7-安全性评估
[0209]
所有安全性评估的时间点提供于soa(表1)中。ae和sae的定义请参见实施例1.8b。就此方案的目的,ms复发(实施例1.6b)免报告为ae,除非符合sae的标准。将非严重的ms复
发收集于特别的ecrf页面并分析作为一效用指标。在ms复发评估后,(实施例1.6b),结论不符合ms复发标准的事件报告为ae。实施例1.7a-身体检查
[0210]
完整的身体检查包括,最低限度,一般外型、头和颈、腹部、淋巴结、皮肤(出血征象,包括瘀伤、瘀点红斑)、心血管、呼吸、胃肠、肌肉骨骼和神经系统的评估。亦测量和纪录身高和体重。简要的身体检查包括,最低限度,皮肤、肺、心血管系统和腹部(肝和脾)的评估。研究者应特别注意与先前严重疾病有关的临床征状。任何新的发现或先前发现的恶化应报告为一新的ae。soa(表1)提供了身体检查的时程。实施例1.7b-生命征象
[0211]
评估温度、脉搏率、呼吸率和血压。整个研究就温度测量应使用相同的方法。以一全自动装置评估坐姿和仰卧时的血压和脉搏测量值。就相同的参与者在整个研究中应使用相同姿势的测量值。仅在无法使用全自动装置时才使用手动技术。在血压测量前应避免咖啡因饮料。血压和脉搏测量应在参与者以安静坐姿无分心下(例如,电视、手机),至少休息5分钟再进行。生命征象(在实验室检测的血液采集前进行)由1脉搏、3血压测量(以至少1分钟间隔记录3个连续血压读数)和呼吸率所组成。记录3个血压读数的平均。实施例1.7c-心电图
[0212]
如soa(表1)所述使用自动计算心率和测量pr、qrs、qt及qtc间隔的ecg机器得到单一的十二导程ecg。需要至少一个较长的心率监测记录作为各ecg检测的部分。ecg由心脏专科医师审查用以确认异常和临床评估。qtc退出标准和任何可能需要的另外qtc读数请参照实施例1.4e。实施例1.7d-临床安全性实验室评估
[0213]
所进行的临床实验室检测列表以及时间和频率的soa(表1)请参见实施例1.16。研究者将会审查实验室报告,记录该审查并将任何研究期间发生的临床上有关的变化记录在ecrf的ae章节中。临床上显著的异常实验室发现是与原发疾病无关的发现,除非由研究者判断比预期的参与者症状更严重。在参与本研究期间或在研究介入最后给剂后4周内,应再重复所有具有视为临床上显著异常的实验室检测,直到回到正常值或基线或由研究者或医疗监测者判定不再认为是临床上显著的。若由研究者合理判断在一段时间内这些数值无法回到正常/基线,则需鉴定病因学并通知赞助者。如实施例1.16中所定义,依照实验室手册和soa(表1)来进行所有方案要求的实验室评估。若来自在机构当地实验室进行的非方案所指定的实验室评估的实验室数值需要改变参与者管理或被研究者认为是临床上显著的(例如,sae或ae或剂量调整),则将结果记录在ecrf。实施例1.7e-自杀风险监测
[0214]
btk抑制剂被认为具cns-活性,且因此进行例行的自杀风险监测。使用哥伦比亚自杀严重程度评定量表(c-ssrs)和详尽的抱怨临床评估进行自杀风险评估。任何临床重要的观察和事件报告为ae。c-ssrs是整个研究中用于评估参与者的终生自杀行为和追踪自杀事件的工具。结构性面谈激起自杀念头的记忆,包括具有实际/潜在致命的念头的强烈性、行为和意图。此量表是由研究者或合格的指定者于soa(表1)中所指的时间点来给予。实施例1.8-不良事件和严重不良事件实施例1.8a-特殊的不良事件
[0215]
aesi是特定于赞助商产品或计划的科学和医疗有关的ae(严重或非严重),需要由研究员持续监测并立即通知赞助者。这些事件可能需要进一步调查以便于找出其特性及了解这些事件。在研究期间可通过方案变更加入、修改或移除特殊的不良事件。-急性超敏反应/过敏性反应-进入研究的女性参与者怀孕以及进入imp/nimp研究的男性参与者的女性伴侣怀孕;
○
进入临床研究的女性参与者怀孕或进入临床研究的男性参与者的女性伴侣怀孕。仅在满足其中一项严重标准时,才有作为sae的资格(参见实施例1.8b)。在女性参与者怀孕的事件中,应中断imp。强制追踪女性参与者怀孕以及男性参与者的女性伴侣怀孕,直到确定结果(参见实施例1.17)-imp/nimp的有症状(严重或非严重)过量给剂:imp/nimp的过量给剂(意外或故意)是因研究者怀疑或由参与者主动通报的事件(并非以全身性药片计数为准)并定义为在预计的治疗期间内至少二倍预计剂量,根据试验药物调整。请注意,无症状过量给剂报告为一标准ae。-alt增加:任何alt增加>3 x uln。其他特定项目的aesi
[0216]
ecg的qtc观察≥500ms或由心脏专科医师确认的临床上显著的心律不整(例如,心房颤动和心房扑动),包括严重感染,尤其是任何机会性感染;重大出血事件,包括在重要区域或器官的有症状出血,例如cns或眼内出血造成一sae;血小板减少症血小板计数<100x109/l。
[0217]
ae是由参与者报告的(或,若适当,由照顾者、代理人或参与者的合法授权代表)。
[0218]
研究者或合格的指定者负责检测、记录及记录符合ae或sae定义的事件并持续追踪严重的、被视为与研究介入或研究程序相关的,或造成参与者中断研究介入的ae(参见实施例1.4e)。
[0219]
ae或sae的定义请参见实施例1.8b。实施例1.8b-不良事件:记录、评估、追踪和报告的定义和程序
[0220]
不良事件(ae):ae是发生在参与者或临床研究参与者的任何不欲的医疗,暂时与使用研究介入有关,无论是否被认为与研究介入有关。ae因此可暂时与使用研究介入有关,任何不利的和非故意的征象(包括异常的实验室数值)、征候或疾病(新的或恶化)。
[0221]
符合ae定义的事件:-任何异常的实验室检测结果(例如,血液学、临床化学或尿液分析)或其他安全性评估(例如,ecg、心脏扫描、生命征象测量),包括与基线相比更恶化,在研究者的医疗和科学判断下认为是临床上显著的(亦即,与原发疾病的进程不相关)。-慢性或间歇性先前存在的症状恶化,包括症状的频率和/或强烈度增加。-在研究介入给药后检测或诊断出新的症状,即使可能在本研究开始之前已存在。-疑似药物-药物交互作用的征象、征候或临床后遗症。-疑似研究介入或并用药物过量给剂的征象、征候或临床后遗症。
[0222]
本身缺乏效用或无预期的药理学作用为报告为一ae或sae,但放入效用评估中。
[0223]
不符合ae定义的事件:-与原发疾病有关的任何临床上显著异常的实验室数据或其他异常的安全性评估,除非由研究者判断参与者症状比预期的更严重-研究的疾病/病症或,或所研究疾病/病症的预期的进展、征象或症候,除非参与者症状比预期的更严重。-医疗或手术过程(例如,内视镜或阑尾切除术):使该过程成为ae的症状-其中未发生不欲的医疗情事的状况(社交和/或方便住院).
‑‑
研究开始时存在或检测到的先前存在疾病或症状的预期日常波动无恶化。
[0224]
若一事件依照上述定义并非为ae,则其亦不可能为sae,即使符合严重症状(例如,因本研究的疾病的征象/征候住院,由于疾病进展而死亡)。
[0225]
严重不良事件(sae):sae为在任何剂量时的任何不欲的医疗情事:a)导致死亡;b)威胁生命(术语“威胁生命”是指其中在事件/反应的时间下参与者处于死亡风险的事件/反应;其并非指假设性若更严重的话可能造成死亡);c)需要住院或造成原本住院时间延长(一般而言,住院意味着该参与者被留在(通常涉及至少停留一夜)医院或急诊室观察和/或治疗,其在医师室或门诊可能不适当。住院期间发生的并发症为ae。若一并发症延长了住院时间或满足任何严重标准,则该事件为一严重事件。当质疑是否要或必要“住院”时,则此ae应视为严重的。选择性治疗与基线相比未恶化的先前症状的住院不能视为ae);d)造成持续的失能/无能(术语失能是指一个人进行正常生活功能的能力被实质破坏。此定义并不希望包括医疗重要性相当小的经验,例如可能干扰或阻碍日常生活功能,但不构成实质上破坏的简单头痛、恶心、呕吐、腹泻、流感和意外创伤(例如,脚踝扭伤));e)先天性异常/先天缺陷;f)其他的状况,例如可能无立即的生命威胁或造成死亡或住院的重要医疗事件,但可能危害参与者或可能需要医疗或手术介入用以防止列于上述定义中的其中一项结果。这些事件通常应为被视为严重的。这些事件的实施例包括侵袭性或恶性癌症、急诊室的加护治疗或家中支气管痉挛、血液恶质病或不会导致住院的抽搐,或发生药物依赖或药物成瘾。
[0226]
ae和/或sae的记录和追踪
[0227]
ae和sae记录:当ae/sae发生时,需审查所有与事件有关的文件记录(例如,医院病程记录、实验室报告和诊断报告)并在ecrf中记录所有相关的ae/sae信息。研究者将参与者医疗记录副本取代ae/sae ecrf页面寄给赞助者代表是不可行的。可能需要提交医疗记录作为额外的sae和aesi报告的资料。在这种情况下其为经匿名化代替参与者的姓名并签署上本研究的参与者编号。可能有赞助者要求特定案例的医疗记录副本的情况。在此情况下,所有的参与者身分标识,参与者编号除外,在提交给自助者之前于医疗记录副本上删除。研究者试图以征象、征候和/或临床数据为基础,建立此事件的诊断。只要有可能,将此诊断(非个别的征象/症候)记录为ae/sae。
[0228]
程度评估:评估研究期间报告ae/sae的程度并分派归属于下列其中一项:-轻度:参与者容易耐受,造成最小程度的不适且不会干扰日常活动的事件。-中度:造成充分的不适并干扰正常的日常活动的事件。-重度:阻碍正常的日常活动的事件。评定为严重的ae不应与sae混淆。重度是用来评比事件强度的项目;ae和sae二者皆可以重度来评定。
[0229]
当一事件符合至少一项sae定义中所述的预定义结果时,而非评比为重度时,则其定义为“严重的”。
[0230]
因果关系评估:研究者有义务评估研究介入和各个ae/sae发生之间的关系。“合理的可能性”的关系为传达有事实、证据和/或论据建议因果关系,而非不能排除关系。研究者使用临床判断来决定此关系。应考虑及调查不明确的原因,例如原发疾病、并用治疗和其他风险因子,以及暂时的事件与研究介入施用的关系。研究者亦就市售产品,以其评估查阅研究者手册(ib)和/或产品信息。
[0231]
就各ae/sae,研究者将其已审查的ae/sae和已提供的因果关系评估记录在医疗记录。可能会有其中sae发生,而研究者仅具有极少的信息结论于初始报告中报告给赞助者的情况。然而,在初始传送sae数据给赞助者之前,研究者始终评估每个事件的因果关系是非常重要的。研究者可能依据追踪信息改变其因果关系的观点并寄发一份具有更新因果关系评估的sae追踪报告。因果关系评定是在决定监管报告要求时所使用的其中一项标准。
[0232]
ae和sae的追踪:当医学上显示或监测小组代表要求解释尽可能完整的ae或sae的性质和/或因果关系时,研究者有义务进行或安排进行补充测量和/或评估。此项应包括额外的实验室检测或研究、组织病理学检查或咨询其他健康照护专家。新的或更新的信息记录在原始完整的ecrf中。若参与者在参与期间或在公认的追踪期期间死亡,则研究者向赞助者代表提供包括组织病理学的任何死后报告副本。新的或更新的信息为记录在原始完整的ecrf中。研究者在接到资料的24小时内,将任何更新的sae数据提交给赞助者。
[0233]
sae报告:经由电子数据收集工具将sae报告给赞助者。sae报告给赞助者的主要机制是电子数据收集工具。若超过24小时无法取得电子系统,则于研究处使用书面sae数据收集工具(参见本文)。研究处应在可取得电子系统时尽快将sae数据键入。在特定地点完成本研究后,应让电子数据收集工具脱机以防止新的数据进入或改变现有的数据。若研究地点在电子数据收集工具脱机后收到研究参与者新的sae报告或收到更新的先前报告的sae数据,则该研究处可将此资料以书面sae形式报告(参见实施例1.8c)或以电话报告给赞助者。
[0234]
经由个案报告表(crf)将sae报告给赞助者:传真传送sae报告crf是将此数据传送给赞助商的优选方法。在极少数的情况或在缺乏传真设备时,以电话通知及以隔夜电邮或快递寄送sae数据收集工具的副本是可接受的。最初的电话通知不能取代研究者必须在指定的报告时间框架内完成和签署sae crf。实施例1.8c-收集ae和sae数据的时间和频率
[0235]
所有的ae(包括sae)是在soa(表1)所述的时间点从签署icf直到eot收集。如实施例1.8b所示,在24小时内记录所有的sae和aesi并报告给赞助者或指定者。研究者将更新的sae数据在其可取得的24小时内提交给赞助者。在研究参与的结论后,研究者并无义务主动找寻ae或sae。然而,若研究者得知任何sae,包括死亡,在参与者退出研究后的任何时间,及研究者认为该事件合理地与研究介入或参与研究相关,则研究者须尽速通知赞助者。记录、评估和评定ae与sae的因果关系以及完成和传送sae报告的程序提供于实施例1.8b中。实施例1.8d-检测ae和sae的方法
[0236]
在检测ae和/或sae时须小心不要导入偏差。参与者的开放式和非引导式口头询问
为优选查询ae事件的方法。实施例1.8e-追踪ae和sae
[0237]
最初的ae/sae报告后,研究者需要在后续的返诊/联络时主动追踪各个参与者。在预先指定的研究结束日期,追踪所有的sae和非严重aesi(如实施例1.8b中所定义)直到解决、稳定,该事件另有解释,或该参与者失访(如实施例1.4e3中所定义)。实施例1.8f-怀孕
[0238]
在研究介入开始后及直到本研究的最后返诊,收集所有女性参与者和男性参与者的女性伴侣怀孕的详情。若有报告怀孕,则研究者在得知怀孕的24小时内通知赞助者而所述程序概述于实施例1.17中。异常怀孕结果(例如,主动堕胎、胎儿死亡、死胎、先天性异常、子宫外孕)视为sae。实施例1.8g-心血管和死亡事件
[0239]
在本研究中心房颤动,心房扑动,qtc时测值≥500ms,或其他临床上显著的心律不整为aesi且进行速报给赞助者。依标准的安全性报告和安全性监督施行(包括idmc的资料审查),报告所有的心血管事件。进行集中ecg审查以确保ecg评估之一致性。依标准的sae报告规则报告死亡事件,用以厘清死亡原因及报告致死事件为sae的诊断。实施例1.8h-多发性硬化症复发报告
[0240]
由实施例1.6b中描述的评估所测定多发性硬化症复发,如同所有的效用指标,免除报告是ae,除非符合sae的定义。ms复发的住院,若例行性在研究处进行(例如,高剂量iv甲基普赖松(methylprednisolone)),就本研究并未视为严重标准。根据一般的安全性报告规则,其他未符合ms复发定义的神经征候的恶化报告为ae。实施例1.8i-核磁共振成像的安全性事件报告
[0241]
核磁共振成像扫描需要于当地就任何非ms病理做审查。若有这些事件发现,则提供mri报告给研究者作为适当的安全性报告。当可取得时,作为此mri发现的原因的病理诊断或该发现本身为报告为ae直到诊断明确。mri上的多发性硬化症发现不需要报告,除非所述发现实际是异常的且因而是不同的安全性事件。实施例1.9-过量给剂的治疗
[0242]
赞助者不推荐特定的过量给剂治疗。在过量给剂的事件中,研究者应:-立即联络医疗监察人。-密切监控该参与者的任何ae/sae及实验室检测异常直到不再全身性检测到研究介入及活动性结束(至少9天)。-若可能从最后剂量的研究介入日期的1天内得到血浆样本进行pk分析,或若医疗监察人要求则在之后(视个案情况而定来决定)。-将超过剂量的数量以及过量给剂的持续时间记录在ecrf中。
[0243]
有关剂量中断或调整由研究者以参与者的临床评估为基准咨询医疗监察人做决定。实施例1.10-药物动力学实施例1.10a-采样时间
[0244]
就二群组的所有参与者,在第1、4、8、12和16周期间的返诊时于给剂后1小时(
±
0.5小时)收集btk抑制剂pk分析的样本。就二群组的所有参与者,在第4和12周期间的返诊
时于给剂后3小时(
±
0.5小时)收集另外的pk样本。将pk采样前最新近的餐食资料标注在ecrf中。实施例1.10b-药物动力学处理程序
[0245]
就各pk样本收集总计2ml的血液。每个参与者pk的血液总量和本研究中所采集的总样本数为如表6所示。表6-每个参与者的血液容量和总样本数实施例1.10c-生物分析法
[0246]
以已验证的lc/ms法分析btk抑制剂。实施例1.10d-pk参数
[0247]
使用叙述统计学报告imp摄取后于选择时间的btk抑制剂浓度。使用群族pk法估算另外的pk参数,例如c
max
、t
max
和稳态时的auc。实施例1.11-药物动力学实施例1.11a-取样时间
[0248]
将收集用于pbmc的静脉血液样本,用于测量基线时及第12和16周期间返诊时btk抑制剂给剂的给剂后1小时(
±
0.5小时)的淋巴细胞亚群分析btk占有率(作为生物标记次研究的部分)。实施例1.11b-用于药效动力学参数的生物分析法
[0249]
从全血制备外围血液单核细胞,用于测定btk占有率。使用描述统计制作在所选时间点的btk占有百分比报告。实施例1.12-药物基因学
[0250]
从同意参加本研究的基因分析部分的参与者收集6ml血液样本进行dna分离。不希望参加基因研究的参与者仍可参与本研究。收集样本用来调查药物代谢酶和/或药物转运子的等位基因变体作为与btk抑制剂的pk和pd变异性有关的内在因子(实施例1.18)。
[0251]
在dna萃取失败的情况下,可向参与者要求替代的基因血液样本。实施例1.13-生物标记
[0252]
如soa(表1)所述,从本研究的所有参与者收集血浆和血清样本用于生物标记研究的。将所有参与者的样本进行神经元丝轻链和几丁质酶3-类蛋白1蛋白及免疫球蛋白量检测,用以评估其与观察到的临床反应的关联性。就其能将样本快速送至中央实验室进行处理的能力所选择的地点的所有参与者,亦收集用于pbmc分离的血液样本。使用外围血液单核细胞样本进行btk受体占有率评估(实施例1.11b),研究期间内选择的淋巴细胞亚群分析,以及其他可能的生物标记。就所有这些样本大约抽取50ml的血液。实施例1.14-统计考虑
实施例1.14a-统计假设
[0253]
本研究的主要目标是在12周的btk抑制剂治疗结束后,以主要终点(脑部mri所检测的新的gd-增强的t1-高信号病灶)为基准,评估剂量反应关系。就主要终点,虚无假设是平的、无剂量反应曲线而对立假设则具有剂量反应信号。实施例1.14b-样本大小决定
[0254]
本研究具有120位参与者平均随机分配至2个群组的4个btk抑制剂剂量其中之一(第1和第2群组各60位参与者)。第1和第2群组代表不同的治疗顺序,且参与者以遮盲的方式各自交叉服用btk抑制剂或安慰剂。
[0255]
第2群组的60位参与者以4周安慰剂导入试验开始,其以在安慰剂治疗的12周期间,固定的每月平均新的gd-增强的t1-高信号病灶数的假设为基础,用作为主要终点分析的安慰剂数据。假设15%的参与者在12周的btk抑制剂结束时无主要终点,使用2步骤mcp-mod以6个预定义剂量反应曲线(2个e
max
模型,1个二次模型,1个线性模型和1个指数模型),105位参与者(每种btk抑制剂剂量26位)具有至少83%的85%最大降低的检测力。此计算假设离差参数2,在第2群组中4周安慰剂和12周btk抑制剂之间的测量值范围从-0.9至0.9的参与者间相关性,及安慰剂在第4周新的gd-增强的t1-高信号病灶平均数目≥1。此检测力使用comprehensive r archive network(cran)的包装剂量探索(package dose finding)(bornkamp b,pinheiro j,bretz f.package
‘
dosefinding’,january 4,2018),使用6个考虑剂量反应模型的候选曲线,在负二项回归框架内来计算。实施例1.14c-分析群族
[0256]
就分析的目的,下列群组为如表7中所示加以定义:表7-分析群族实施例1.14d-统计分析
[0257]
效用分析
[0258]
主要分析:以建模技术的2步骤多重比较法(mcp-mod)于修改的治疗意向(mitt)群族中评估btk抑制剂与主要终点(12-周的btk抑制剂治疗结束时脑部mri所检测的新的gd-增强的t1-高信号病灶数)的剂量反应关系的主要目标。此方法的第一步骤为检测控制第1型错误的方法中的效用信号(与平的、无剂量反应曲线的虚无假设相比较)。就说明剂量反
应形状的不确定性,考虑6个候选模型来涵盖各式各样和可能的剂量反应样貌:2个e
max
模型(ed
50
=10mg,ed
50
=30mg),线性模型,二次模型、逻辑模型和指数模型。第二步骤是剂量反应曲线的剂量估算,其限制条件为在第一步骤中建立效用信号。
[0259]
使用带有基线gd-增强的t1-高信号病灶数、治疗和群组(第1群组或第2群组)的共变项的负二项回归模型,评估各4个剂量组在12-周的btk抑制剂治疗结束及4周安慰剂结束时的平均新的gd-增强的t1-高信号病灶数。在假若参与者于12周期间接受安慰剂,gd-增强的t1-高信号病灶形成率固定的假设下,利用第2群组的4-周的随机分配后安慰剂数据(亦即第2群组的第4周数据)作为分析中第12周时的安慰剂数据。第2群组的参与者贡献安慰剂数据(第4周)以及4个btk抑制剂剂量的数据(第16周)。因此,为了说明第2群组中4-周安慰剂期和后续12-周btk抑制剂治疗期测量值之间的可能相关性,使用广义估计方程式(gee)法来拟合负二项回归模型,经由sas proc genmod中重复的叙述来说明参与者间的相关性。将负的平均病灶数的对数变换代入mcp-mod法。在主要终点12周的btk抑制剂治疗结束时,将平的剂量反应曲线(亦即无剂量反应关系)的虚无假设共同对各6个候选剂量反应模型以控制族系误差率(family wise error rate)在2-侧α=0.05的对比试验进行评估。若步骤1产生显著的结果,则使用广义的赤池信息量准则(generalized akaike information criterion)(aic)从6个预定义候选模型中选定最佳的拟合模型。
[0260]
主要分析以第1和第2群组就各btk抑制剂剂量的合并数据为基准(亦即,就第1群组为第12周而第2群组为第16周的新的gd-增强的t1-高信号病灶数的数据)。若需要,第1和第2群组的数据亦可分开探讨。
[0261]
次要指标的分析:就12周btk抑制剂治疗结束时gd-增强的t1-高信号病灶数的次要终点,使用类似的负二项回归模型和mcp-mod法。因对于gd-增强的t1-高信号病灶数,在安慰剂下12周期间固定的病灶形成率的假设是合理的,因此在说明病患内的相关性时,使用主要终点所利用的相同方法,通过使用第2群组第4周的数据作为第12周安慰剂的数据。提供各4个btk抑制剂剂量在该段时间内的描述性概括统计。
[0262]
就新的或扩大的t2病灶数,提供各4个btk抑制剂剂量随时间变化(4、8、12和16周)的描述性概括统计。再者,若由第2群组第4周数据推断第12周安慰剂数据是实际上合理的时,则探索类似的mcp-mod法。
[0263]
主要效用分析以mitt群族为基础。就与基线相比的变化所评估的终点,基线值定义为在研究介入的第1剂开始前,在随机分配诊察时(第1天)或之前所收集的最后测量值。
[0264]
将第1和第2群组的数据组合用于主要分析(亦即第1群组第12周和第2群组的新的gd-增强的t1-高信号病灶的数据)。就各群组,若适当,汇总随时间变化的描述统计(第4、8、12和16周)。第1群组的汇总包括在12周的btk抑制剂治疗后的4周安慰剂期的描述统计。另外的效用分析描述于sap中。表8-效用分析
[0265]
安全性分析
[0266]
于安全性群族上进行所有的安全性分析。所有安全性概括是描述性的。并无在安全数据上进行统计显著性试验。安全性终点为如表9中所述。
[0267]
基线值一般定义为在第一次给予随机分配的研究介入之前,最后可取得的数值。随机分配后(其中第2群组的参与者为接受安慰剂),前面4周的安全性数据以btk抑制剂和安慰剂汇总。分开汇总第1群组4-周安慰剂期间的安全性数据(亦即4周)并显示btk抑制剂剂量组和全部数据。就btk抑制剂治疗安全性数据,提供剂量组、btk抑制剂的时间及全部的汇总。
[0268]
就安全性变量,定义下列观察期并用于ae分类,测定经治疗的pcsa值,以及最后经治疗的实验室和生命征象参数的数值。治疗前的时期定义为签署icf至第一次给予随机分配研究介入的时间。就定义“治疗中出现”的目的,治疗进行期定义为从第一次给予随机分配研究介入直到最后研究返诊的时间。治疗期进一步定义为:
‑“
1至4周期间”定义为从第一次给予随机分配研究治疗至给予第4周的研究治疗的时间。就第1群组,为btk抑制剂治疗4周而就第2群组为安慰剂治疗4周。
‑“
btk抑制剂治疗期”就第1群组,定义为第1至12周,就第2群组为第4至16周。注意:就第1群组的参与者,1至4周期间亦包括在12周的btk抑制剂治疗期的中。
‑“
安慰剂/btk抑制剂给剂期之后”就第1群组定义为第12至16周。其为12周的btk抑制剂治疗之后的4周安慰剂治疗。
[0269]
ae分析聚焦在治疗中出现的不良事件(teae)。治疗前ae定义为在治疗前发生、恶化或变严重的ae。治疗中出现的ae(teae)定义为在治疗期间发生、恶化或变严重的ae。
[0270]
下列定义适用于实验室参数、ecg和生命征象结果:-可能的临床上显著异常(pcsa)值为义为由研究者根据预定义的标准/阈值以文献回顾为基准认为是医疗上重要的异常值且为由赞助者就临床实验室检测和生命征象所定义。-可能的临床上显著异常标准决定,参与者在治疗期间具有至少1次pcsa,包括非时程内的或重复的评估。所有这些参与者的人数为治疗中pcsa百分比的分子。
[0271]
表9-安全性分析
[0272]
个别的pk浓度以返诊描述性汇总。另外的pk参数如实施例1.10d所述,以及群族pk和pd分析呈现于单独的文件中。实施例1.15-期中分析
[0273]
若由于招募比预期的更慢而实际需要时,当至少44位参与者已完成16周的研究时(12周的btk抑制剂治疗和4周的安慰剂),可进行一期中分析。若试验招募快速则不进行期中分析,因为期中分析可能太接近有价值的最终分析的时间(亦即,少于3至4个月)。期中分析的目的应是探索效用信号和优化第3期研究规画。操作文件预先指定进行期中分析的条件(例如,招募率)以及在sap定案前决定是否进行期中分析。若进行期中分析,则探讨相较于安慰剂(使用第2群组4-周随机分配后安慰剂剂数据)的新的gd-增强的t1-高信号病灶数(仅在60mg组或60和30mg组合组中进行),以及可能的剂量反应曲线。期中分析应由未遮盲独立的统计员来进行。因为本研究不会基于此可能的探索性期中分析因效用声明而提早停止,所以若进行期中分析,在最终分析时不会做α调整。假如欲进行期中分析,则sap更详尽地描述所规划的1个期中分析
[0274]
使用idmc监控本研究的安全性。实施例1.16-临床时实验室检测
[0275]
临床实验室检测的详情提供于表10中。如研究者决定有必要或当地法规要求,则
在研究期间的任何时间进行额外的检测。表10-协定要求的安全性实验室评估
实施例1.17-避孕指南和收集怀孕信息
[0276]
具生育能力的妇女(wocbp):除非永久不育,否则妇女在初潮后及直到停经后为止,视为是有生育力的。
[0277]
下列项目中的妇女不视为wocbp:1)初经前期2)具有下列其中1项的停经前女性:有记录的子宫切除、有记录的双边输卵管切除,有记录的双边卵巢切除。3)停经后女性-停经后的状态定义为无替代的医疗因素下12个月无月经。在停经后范围内高量的fsh可用于确认无使用荷尔蒙避孕或荷尔蒙替代治疗(hrt)的妇女的停经后状态。然而,缺乏闭经12个月,单一的fsh测量并不够。-进行hrt及停经状态有疑问的女性需要使用其中一种非雌激素荷尔蒙高效避孕法,若其在研究期间希望持续hrt的话。否则在纳入研究之前其必须中断hrt以确认停经后状态。
[0278]
避孕指南
●
男性参与者
○
若从纳入到最后剂量的研究介入后高达3个月,具有生育能力的女性伴侣的男性参与者同意下列其中一项,则有资格参加:
■
禁断阴茎-阴道性交作为惯常和偏爱的生活方式(长期和持续性地戒除),并同意保持禁欲
■
当与目前未怀孕、有生育能力的妇女具有阴茎-阴道性交时,同意使用男用避孕套加上伴侣使用如表11所示每年失败率<1%的避孕法
○
此外,男性参与者在研究期间及在最后剂量的研究介入之后6个月必须避免捐赠精子
○
具有怀孕或哺乳伴侣的男性参与者必须同意维持禁断阴茎-阴道性交或在每次阴茎进入期间使用避孕套直到最后给剂后3个月。
●
女性参与者
○
因为尚未进行btk抑制剂的决定性的生殖毒性研究,因此研究者在本临床试验的wocbp暴露期间直接采取适当的预防措施。若其同意从纳入到最后研究给剂后高达2个月,使用双重避孕法,包括持续和正确地如表9中所述的高效避孕法,则具生育能力的女性参与者有资格参加。此外,wocbp在研究期间及在最后剂量的研究介入之后2个月必须避免捐赠卵子。表11.高效避孕法
[0279]
怀孕检测:仅在确认月经期和高敏感血清怀孕检测阴性之后,才会纳入wocbp。在介入期间和最后剂量的研究介入后1个月以及如当地要求,则进行另外的怀孕检测。当月经没来或除此之外怀疑怀孕时,则进行怀孕检测
[0280]
收集怀孕信息:
[0281]
男性参与者的伴侣怀孕-研究者力尽力于男性参与者在本研究中时收集任何男性参与者的女性伴侣怀孕的信息。此项仅适用于接受btk抑制剂的男性参与者。在从怀孕的女性伴侣直接得到必要的签署过的知情同意书后,研究者在适当的表格上记录怀孕数据并在得知该伴侣怀孕的24小时内将该信息提交给赞助者。亦追踪该女性伴侣最终的怀孕结果。将母亲和小孩的状况信息转发给赞助者。一般而言,追踪不会超过预产期后的6至8周。报告任何怀孕终止,无论胎儿的状况(有或无异常现象)或处置适应症。
[0282]
女性参与者怀孕-研究者于女性参与者在本研究中时收集任何怀孕的的女性参与者的怀孕信息。将怀孕数据记录在适当的表格上并在得知参与者怀孕的24小时内提交给赞助者。追踪该参与者最终的怀孕结果。研究者收集追踪参与者和新生儿的数据并将该数据转发给赞助者。一般而言,追踪不需要超过预产期之后的6至8周。报告任何怀孕终止,无论胎儿的状况(有或无异常现象)或处置适应症。任何怀孕并发症或选择性终止怀孕为报告为ae或sae。自发性堕胎通常认为是sae并将按此报告。任何与sae有关的研究后怀孕,研究者合理考虑与研究介入有关,则报告给赞助者。在研究者无义务主动寻查之前参与者的此项数据的同时,研究者可能经由自动报告而得知sae。任何在参与本研究时怀孕的女性参与者则中断研究介入并退出本研究。实施例1.18-基因学
[0283]
使用/分析dna
[0284]
基因变异可能影响参与者对研究介入的反应,容易罹患疾病,以及疾病的严重度和进程。各种对研究介入的反应可能由于影响药物吸收、分布、代谢及排泄的基因决定子;药物作用机制;疾病病因学;和/或所治疗疾病的分子亚型。因此,在当地法规和irb/iec允许下,从同意的参与者收集血液样本供dna分析。
[0285]
使用dna样本进行有关本研究介入或ms或相关疾病的研究。其亦可用来开发试验/分析包括与研究介入和适应症有关的诊断试验。基因研究可能包括一个或多个候选基因的分析或整个基因组基因标记的分析(若需要)。分析dna样本用以调查药物代谢酶的等位基因变体和/或药物转运子作为与btk抑制剂的pk或pd变异性有关的内在因子。若假设另外的分析可能帮助进一步了解临床数据,则可进行另外的分析。可分析样本作为涉及btk抑制剂反应的基因因子的多重研究评估的部分或了解研究疾病或相关症状的此类研究介入。实施例1.19-禁用的示例药物列表
[0286]
在研究期间由于与p450-介导的代谢相互作用(其为强力的cyp3a或cyp2c8肝酶的诱发剂或抑制剂),可能改变btk抑制剂动力学,所以不应服用下列药物(依华盛顿大学药物交互作用数据库程序列表(www.druginteractioninfo.org)。请注意,所提供的列表并不详尽且对于意欲并用药物的产品信息应咨询过。表12-可能改变btk抑制剂代谢的示例药物列表
表13-可能经由降低胃酸影响btk抑制剂血浆暴露的示例药物列表表14-缩写
实施例2:复发型多发性硬化症中找寻btk抑制剂剂量和安全性研究结果
[0287]
我们于文中提供了如实施例1中所述的剂量探索和安全性研究的结果。测定了btk抑制剂降低新的活动性脑病灶数,包括新的钆(gd)-增强的t1-高信号病灶数的剂量反应关系。亦以造影测量评估,测量新的或扩大的t2病灶数及gd-增强的t1-高信号病灶总数,评估btk抑制剂对疾病活动性的效用。亦评估btk抑制剂剂量反应的安全性和耐受性。
[0288]
如实施例1中所述,本研究是在欧洲和北美具有总计40个活动地点的多中心研究。所有的参与者在交叉前使用交互式语音/网络回复系统集中分配至8臂中的其中之一(2个群组的各自的4个剂量组以相同比例,开始btk抑制剂(第1群组)或安慰剂(第2群组)期。
[0289]
在各群组内,参与者以遮盲的方式随机平均分配至4个btk抑制剂剂量(每天一次5、15、30或60mg)的其中之一。
[0290]
第1群组:参与者在前面12周接受其中1个btk抑制剂剂量,然后交叉进行4周安慰剂。
[0291]
第2群组:参与者在前面4周接受安慰剂,然后交叉进行12周的其中一个btk抑制剂剂量。
[0292]
所有的脑部扫描在独立的中央设施由1或多位对治疗不知情的评定遮盲放射线医师来审查和解释,由此避免偏差并确保标准化的终点评估。
[0293]
诊断和纳入标准:年龄18至55岁的参与者,根据2017修订版的麦当劳诊断标准经诊断患有rms且在前1年内具有至少1次记录的复发在前2年内or≥2记录的复发,在筛选前6个月内mri扫瞄or≥1活动性gd-增强的脑病灶。
[0294]
主要和次要关键终点
[0295]
效用:
●
主要:在12周的btk抑制剂治疗结束后如脑部mri所检测,新的钆(gd)-增强的t1高信号病灶数。
[0296]
次要:
●
12周的btk抑制剂治疗结束时新的或扩大的t2病灶数;
●
12周的btk抑制剂治疗结束时gd-增强的t1-高信号病灶数
[0297]
安全性:
●
不良事件(ae),严重不良事件(sae),实验室检测的可能的临床上显著异常,心电图(ecg),或研究期期间生命征象
[0298]
统计方法:
[0299]
主要终点的分析:
[0300]
主要分析以汇总的第1和第2群组的各btk抑制剂剂量的数据为基础(亦即,第1群组为新的gd-增强的t1-高信号病灶数的第12周的数据而第2群组为第16周的数据)。
[0301]
以建模技术的2步骤多重比较法(mcp-mod)于修改的治疗意向(mitt)群族中评估btk抑制剂与主要终点(12-周的btk抑制剂治疗结束时脑部mri所检测的新的gd-增强的t1-高信号病灶数目)的剂量反应关系的主要目标。此方法的第一步骤为检测控制第1型错误的方法中的效用信号(与平的、无剂量反应曲线的虚无假设相比较)。就说明剂量反应形状的不确定性,考虑6个候选模型来涵盖各式各样和可能的剂量反应样貌:2个e
max
模型(ed
50
=10mg,ed
50
=30mg),线性膜性,二次模型、逻辑模型和指数模型。因为在第一步骤中建立了效用信号,所以第二步骤为估算剂量反应曲线。
[0302]
在mcp-mod步骤1中,使用具有共变项的负二项回归模型,就基线gd-增强的t1-高信号病灶活动性(有/无)及治疗,评估各4个剂量组在12周的btk抑制剂治疗结束及4周安慰剂结束时的平均新的gd-增强的t1-高信号病灶数。若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。在假若参与者于12周期间接受安慰剂,gd-增强的t1-高信号病灶形成率为固定的假设下,利用第2群组的4周的随机分配后安慰剂数据(亦即第2群组的第4周数据)作为分析中第12周时的安慰剂数据。第2群组的参与者贡献安慰剂数据(第4周)以及4个btk抑制剂剂量的数据(第16周)。第1群组的4周安慰剂导出数据并未纳入本分析中。因此,为了说明第2群组中4周安慰剂期和后续12周btk抑制剂治疗期测量值之间的可能相关性,为使用广义估计方程式(gee)法来拟合负二项回归模型,通过使用sas proc genmod中“重复的”叙述来说明参与者内的相关性。将负的平均病灶数的对数变换代入mcp-mod法。在主要终点12周的btk抑制剂治疗结束时,将平的剂量反应曲线(亦即无剂量反应关系)的虚无假设共同对各6个候选剂量反应模型使用控制族系误差率(family wise error rate)在双边α为0.05的对比试验来进行评估。提供全部6个候选模型的检验统计量和调整的p值。
[0303]
在mcp-mod步骤2中,为拟合步骤1中调整p值<0.05的所有候选模型。提供广义的赤池信息量准则(aic)和模型参数。选择具有最小广义aic者作为最佳拟合模型。然后从最终选择的模型估算用于第3期计划的剂量。
[0304]
另外,就各4个btk抑制剂剂量组以上述的负二项回归模型为基础,提供相对于安慰剂组的相对降低的平均gd-增强的t1-高信号病灶数和对应的95%信赖区间(ci)。
[0305]
亦提供各4个btk抑制剂剂量随时间变化的实测的新的gd-增强的t1-高信号病灶数(亦即,就第1群组/第2群组为第4周/第8周,第8周/第12周,及第12周/第16周)和安慰剂组(第2群组第4周)的描述性统计。
[0306]
次要终点分析:
[0307]
就各次要终点,为使用类似的负二项回归模型和mcp-mod法。因为在安慰剂治疗下假设固定的病灶形成率为合理的,因此在说明参与者间的相关性时使用与用于主要终点的
相同方法,亦即使用第2群组中第4周的数据作为第12周的安慰剂数据。提供各4个btk抑制剂剂量组的随时间变化的描述性概括统计。
[0308]
所有的安全性汇总是描述性的且于安全性群族上进行。随机分配后之前面4周的安全性数据(第2群组的参与者接受安慰剂时)以btk抑制剂和安慰剂治疗汇总。btk抑制剂治疗期期间(从第1次btk抑制剂给药)的安全性数据以btk抑制剂剂量组和整体来汇总。
[0309]
群族特性:
[0310]
随机分配130位病患。
[0311]
基线时参与者的人口统计和特性一般在8个治疗臂之间为平衡良好的(2个群组的各自的4个治疗臂)。参与者的中位数年龄为36.3岁(范围:19至55岁)。大多数的参与者为女性(91位;70.0%)。请注意130位参与者中有119位(91.5%)为白人。
[0312]
全部130位参与者经诊断皆患有rms(128位具有复发性rms而2位患有续发进行性ms),其中平均edss得分为2.50,从初次诊断罹病的中位数时间为3.5年,及初次ms征候出现的中位数时间为4.9年。127位(97.7%)参与者在筛选前1年内具有至少1次复发,及61位(46.9%)参与者具有在筛选前1年内1次复发所定义的高活动性疾病(had)以及筛选前6个月内所作的mri中≥1个gd-增强的病灶,或在基线时9个或更多个t2病灶,或筛选前1年内复发2次或更多次)。
[0313]
130位病患中有129位完成治疗期。1位参与者在第12周后因拒绝遵守避孕要求而永久中断治疗。
[0314]
表15提供病患处置的详情。表16a-16b汇总病患的人口统计和基线特性。表17显示各组的暴露持续时间的详情而表18显示各组的剂量的暴露详情。
[0315]
效用结果:
[0316]
主要终点:本研究满足其主要目标,验证了btk抑制剂的剂量反应关系,如在12周治疗后由脑部mri所检测的新的活动性gd-增强的t1-高信号脑病灶数降低所证实。
[0317]
表19提供12周的治疗后对比安慰剂新的gd-增强的t1-高信号脑病灶的相对降量的汇总,亦如图2a中所示。表20显示12周的治疗后新的gd-增强的t1-高信号脑病灶的mcp-mod。如上文统计方法章节中所述,进行mcp-mod评估。
[0318]
图2b显示mcp-mod步骤2用于评估新的gd-增强的t1-高信号脑病灶的预估的剂量反应曲线。为了说明第2群组导入期(run-in period)和治疗期测量值之间的可能相关性,使用广义估计方程式法(gee)来拟合说明参与者内相关性的模型。从负二项回归模型估算btk抑制剂治疗在第12周和安慰剂在第4周的平均病灶数,并说明第2群组中4周安慰剂期和后续12周btk抑制剂治疗期测量值之间的可能相关性。若参与者在mri评估日前的30天内接受全身性皮质类固醇,则将此mri评估从该分析中排除。
[0319]
如图2a和表19中所示,在治疗后12周观察到的平均(sd)新的gd-增强的t1-高信号病灶数在安慰剂组中为1.03(2.50),在btk抑制剂5mg组中为1.39(3.20),在btk抑制剂15mg组中为0.77(1.48),在btk抑制剂30mg组中为0.76(3.31),及在btk抑制剂60mg组中为0.13(0.43)。从针对基线gd-增强的t1-高信号病灶活动性做调整的负二项回归模型,相较于安慰剂,在12周相对的病灶降低量在60mg剂量组中系统计上显著的(85.02%;95%ci[28.02%,96.88%];名义上p-值=0.0178),但在较低剂量组中则否。请注意,在btk抑制剂60mg剂量组中以可评估的mri数据有90.30%的参与者(31位中有28位)在12周的治疗结束
时没有新的gd-增强的t1-高信号病灶。选择指数模型作为最佳拟合剂量反应曲线并如图2b中所示。
[0320]
主要的次要终点:表21提供12周的治疗后对比安慰剂新的或扩大的t2病灶数的相对降量的汇总,亦如图3a中所示。表22显示12周的治疗后新的或扩大的t2病灶数的mcp-mod,亦如图3b中所示。如上文统计方法章节中所述,进行mcp-mod评估。表23提供12周的治疗后对比安慰剂的总t2gd-增强的t1-高信号病灶数的相对降低量的汇总。表24显示12周的治疗后gd-增强的t1-高信号病灶总数的mcp-mod。12周的治疗后就第1群组的btk抑制剂治疗代表第12周,就第2群组的btk抑制剂治疗为第16周,就第2群组安慰剂为第4周。若参与者在mri评估日前的30天内接受全身性皮质类固醇,则将此mri评估从该分析中排除。mcp-mod 1无显著性。
[0321]
如表21和图3a中所示,就次要终点-在12周的btk抑制剂治疗结束时,新的和扩大的t2病灶数,在安慰剂中观察到的平均(sd)为2.12(5.16),在btk抑制剂5mg组中为1.90(3.97),在btk抑制剂15组中为1.32(1.83),在btk抑制剂30mg组中为1.30(4.90),在btk抑制剂60mg组中为0.23(0.62)。相对于安慰剂组,在新的和扩大的t2病灶数经调整的相对降低量上60mg剂量组,不同于其他剂量组,显示统计上显著的(89.34%;95%ci:[68.39%,96.41%];名义上p-值=0.0001)。在btk抑制剂60mg剂量组中以可评估的mri数据有87.1%的参与者(31位中有27位)在12周的治疗结束时没有新的和扩大的t2病灶。选择线性模型作为最佳拟合剂量反应曲线并如中图3b所示。
[0322]
如表23中所示,就次要疗效指数-在12周的btk抑制剂治疗的gd-增强的t1-高信号病灶数,在安慰剂组中观察到的平均(sd)为1.36(3.52),在btk抑制剂5mg组中为1.77(4.10),在btk抑制剂15mg组中为0.87(1.59),在btk抑制剂30mg组中为1.18(4.87),以及在btk抑制剂60mg组中为0.29(0.86)。在任何受试的btk抑制剂剂量中相对于安慰剂并未观察到统计上显著的相对病灶数降低量。然而,相较于安慰剂组(74.6%,59位中有44位),在12周的治疗结束时,观察到btk抑制剂60mg组以可评估的mri数据具有较高百分比的无gd-增强的t1-高信号病灶(87.1%,31位中有27位)。
[0323]
安全性结果:btk抑制剂在12周的治疗期间是耐受良好的。表25a提供1-4周期间治疗中不良事件的概述。表25b提供4-周期间不良事件的概述。表25c提供12-周期间不良事件的概述。表26提供btk抑制剂治疗期中治疗中出现的不良事件的概述。表27提供btk抑制剂治疗期中严重的治疗中出现的不良事件的概述。表28提供1-4周期间治疗中出现的特殊关注不良事件的概述。表29提供btk抑制剂治疗期中治疗中出现的特殊关注不良事件的概述。表30提供在12周治疗期间所有剂量中有2个以上的病患发生不良事件的概述。
[0324]
如表17中所示,就5、15、30和60mg的btk抑制剂组,btk抑制剂暴露的平均持续时间为82.1、81.5、83.6和82.1天,而安慰剂组在前16周期间为28.0天。btk抑制剂整个给剂组的平均暴露时间为82天。
[0325]
在研究中无报告死亡。在第1群组1位以60mg btk抑制剂治疗参与者中报告1件治疗中出现的sae。该事件,发生在32岁ms复发的女性参与者中,为在开始以btk抑制剂治疗后大约8周发生。该参与者具有说话困难及喝饮品时会流口水。在住院纪录中并无报告吞咽困难。该参与者在经历此征候后2天住院,排除中风可能。该事件由研究者评判为严重事件。确认ms复发,持续治疗不中断,且该参与者完成本研究并成功进入长期延伸研究。
[0326]
除了一件在60mg btk抑制剂组中报告的严重teae(上述严重ms复发的sae)之外,所有报告的teae为轻度或中度。
[0327]
无teae导致永久性治疗中断。在安慰剂组和5、15、30和60mg组中在第1至4周中经历teae的参与者比例分别为34.8%、31.3%、18.8%、12.5%和31.3%。在治疗期期间teae的参与者比例在4个btk抑制剂组中为相似的(在5、15、30和60mg btk抑制剂组中分别为57.6%、53.1%、54.5%和50.0%)。
[0328]
在第1至4周中(安慰剂对照)主要soc最常报告的teae(总共>3件)是头痛(安慰剂组中4例,btk抑制剂5mg组中3例,btk抑制剂15mg组中2例,btk抑制剂60mg组中1例),上呼吸道感染(各治疗疗组,包括安慰剂,1例),恶心(安慰剂组中1例,btk抑制剂5mg组中2例,及btk抑制剂30mg组中1例)。
[0329]
如表30中所示,在12-周btk抑制剂治疗期中主要soc最常报告的teae为:头痛(btk抑制剂5mg组中1例,btk抑制剂15mg组中3例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中4例),上呼吸道感染(btk抑制剂5mg组中2例,btk抑制剂15mg组中2例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中1例),鼻咽炎(btk抑制剂5mg组中1例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中3例),背痛(btk抑制剂5mg组中1例,btk抑制剂15mg组中1例,及btk抑制剂30mg组中2例),外周性水肿(btk抑制剂5mg组中2例,及btk抑制剂60mg组中2例),肠胃炎(btk抑制剂5mg组中1例,及btk抑制剂60mg组中2例),呼吸道感染(btk抑制剂15mg组中1例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中1例),肌肉痉挛(btk抑制剂30mg组中1例,及btk抑制剂60mg组中2例),咽喉痛(btk抑制剂5mg组中1例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中1例),脱发(btk抑制剂5mg组中1例,btk抑制剂15mg组中1例,及btk抑制剂60mg组中1例),丙氨酸氨基移转酶增加(btk抑制剂5mg组中1例,btk抑制剂30mg组中1例,及btk抑制剂60mg组中1例)和意外的给剂过量(btk抑制剂60mg组中3例)。3位经历脱发的参与者具有可能应能说明该事件的医疗病史。
[0330]
如表29中所示,在本研究中报告二件aesi(丙氨酸氨基移转酶增加>3 x uln),1件在30mg btk抑制剂治疗期期间及1件在60mg btk抑制剂治疗期期间。在该2例中,肝酶升高是过渡性的,未中断imp,肝酶回到正常值,且该参与者完成本研究。接受60mg的参与者以及报告并发搔痒的事件由研究者评定为轻度而另一件无并发症为判定为中度。1位参与者(60mg组)在筛选和随机分配时具有alt量高于uln(34u/l)(分别为48和50u/l)且在第4周返诊时达到>3xuln量(107u/l)。在第12周alt量逐渐下降,达到正常量(28u/l)。另1位参与者(30mg组)在第8周返诊时具有alt量>3xuln(105u/l),且在4天内回到正常量(32u/l)。2位参与者皆为女性。
[0331]
与生命征象(收缩压,体重),实验室(血红素≤115g/l[男性];≤95g/l[女性],血球比容≤0.37v/v[男性];≤0.32v/v[女性],alt>3 x uln,胆红素>1.5 x uln)及ecg(例如,心跳率<50下/min,心跳率>90下/min,心跳率>100下/min,pr>200msec,qrs>110msec,qtc bazett>450msec,qtc fridericia>450msec)有关的pcsa报告于多重治疗组中,无剂量关系。
[0332]
结论
[0333]
本研究满足其主要目标,验证了btk抑制剂的剂量-反应关系,如在12周治疗后由脑部mri所检测的新的活动性gd-增强的t1-高信号脑病灶数降低所证实,其中相较于安慰
剂,btk抑制剂60mg组具有统计上显著差异;相较于安慰剂,其他btk抑制剂治疗组的差异并非统计上显著的。一致地,亦由12周的60mg btk抑制剂治疗后脑部mri所检测的新的和扩大的t2病灶数下降验证了对疾病活动性的效用,但就5、15和30mgbtk抑制剂剂量则无此效用。然而,无论哪一个受试剂量,数据并未显示12周的btk抑制剂治疗后gd-增强的t1-高信号病灶总数在统计上显著的降低。
[0334]
施用的btk抑制剂剂量与teae数量之间并无直接相关性。在btk抑制剂治疗臂的参与者中观察到最常见的事件(优先项)为头痛、上呼吸道感染和鼻咽炎。在多剂量组中观察到低数量的aesi和pcsa。在本试验中确认并无新风险。
[0335]
这些发现显示,在此剂量范围内的btk抑制剂治疗于复发型ms病患中就低剂量mri病灶为耐受良好且有效的。
[0336]
表15-病患处置
n:该项目的参与者人数,n:随机分配的参与者人数,btki:btk抑制剂注意:百分比是使用随机分配的参与者人数作为分母所计算的。表16a-人口统计和基线特性人口统计和基线时参与者特性-随机分配群族
bmi:身体质量指数,n:该项目的参与者人数,n:随机分配的参与者人数,安慰剂:第2群组4周安慰剂期,btki:btk抑制剂表16b-病患基线特性
edss=扩展失能状况量表;gd=钆;rrms=复发-缓解多发性硬化症。高活动性疾病定义为在筛选前1年内复发1次及在筛选期6个月内进行的mri上≥1个gd-增强的病灶或基线时≥9个t2病灶,或筛选前1年内复发≥2次。数值为平均值(sd),但标注的地方除外;a仅包括第2群组安慰剂臂,其于第4周开始btki治疗;bn=127;cn=32;dn=31。表17-暴露的持续时间:安全性群族暴露于研究药品的程度-安全性群族
n:该项目的参与者人数,n:随机分配的参与者人数,安慰剂:第2群组4-周安慰剂期,btki:btk抑制剂imp暴露的持续时间定义为最后给剂日-第一次给剂日 1天,无论是否有计划之外
之间歇性中断表18-暴露的持续时间:安全性群族暴露的持续时间:安全性群族表19-描述性概括和对比安慰剂的12周治疗后新的gd-增强的t1-高信号病灶的相对降低量
btki:每日btk抑制剂mg*12周的btk抑制剂治疗后就第1群组btk抑制剂治疗代表第12周,就第2群组btk治
疗为第16周,就第2群组安慰剂为第4周。**若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。#就基线gd增强的t1高信号病灶活动性调整负二项式模型(有/无)****p值无多重调整表20-12周治疗后的新的gd-增强的t1高信号病灶的mcp-modbtki:每日btk抑制剂mg*12周的btk抑制剂治疗后就第1群组btk抑制剂治疗代表第12周,就第2群组btk治疗为第16周,就第2群组安慰剂为第4周。注意1:若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。注意2:选择具有最小aic的指数模型。表21-描述性概括和对比安慰剂的12周治疗后新的或扩大的t2-病灶数的相对降低量
btki:每日btk抑制剂mg*12周的btk抑制剂治疗后就第1群组btk抑制剂治疗代表第12周,就第2群组btk治疗为第16周,就第2群组安慰剂为第4周。
**若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。#就基线t2-病灶数活动性调整负二项式模型(有/无)***p值无多重调整表22-12周治疗后的新的和扩大的t2病灶的mcp-modbtki:每日btk抑制剂mg*12周的btk抑制剂治疗后就第1群组btk抑制剂治疗代表第12周,就第2群组btk治疗为第16周,就第2群组安慰剂为第4周。注意1:若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。注意2:选择具有最小aic的线性模型。表23-描述性概括和对比安慰剂的12周治疗后gd-增强的t1-高信号病灶总数的相对降低量*
btki:每日btk抑制剂mg*12周的btk抑制剂治疗后就第1群组btk抑制剂治疗代表第12周,就第2群组btk治疗为第16周,就第2群组安慰剂为第4周。注意1:若参与者在mri评估日之前的30天内接受全身性皮质类固醇,则将该mri评估从分析中排除。#就基线gd增强的t1高信号病灶活动性调整负二项式模型(有/无)***p值无多重调整表24-12周治疗后的gd-增强的t1高信号病灶总数的mcp-mod
表25a-治疗中出现的不良事件的概述-第1至4周teae:治疗中出现的不良事件,sae:严重的不良事件n(%)=具有至少1件teae的参与者人数和百分比注意:安慰剂为第2群组4-周安慰剂期表25b-不良事件的概述-第1至4周
表25c-不良事件的概述-第1至12周表26-治疗中出现的不良事件的概述-btk抑制剂治疗期,安全性群族
teae:治疗中出现的不良事件,sae:严重的不良事件n(%)=具有至少1件teae的参与者人数和百分比表27-严重的治疗中出现的不良事件-btk抑制剂治疗期,安全性群族在btk抑制剂治疗期中主要soc和pt的具有治疗中出现sae的参与者人数(%)-安全性群族sae:严重不良事件,soc:系统器官分类,pt:优先项meddra 22.1n(%)=具有至少1件治疗中出现的sae的参与者人数和百分比注意:表格是根据所有teae的汇总以soc国际上认可顺序和以下降频率分类的pt来分类的表28-特殊关注的治疗中出现的不良事件-第1至4周
表29-特殊关注的治疗中出现的不良事件-btk抑制剂治疗期,安全性群族表30-在12周治疗期间所有的剂量发生于>2位病患中的不良事件
实施例3
[0337]
使用小鼠初代微胶细胞,研究btk的角色。产生小鼠初代微胶细胞
[0338]
收集产后小鼠脑部
[0339]
从c57bl/6产后小鼠的脑部得到微胶细胞的初代培养物。将小鼠以co2麻醉及随后以灭菌过的剪刀斩首。收取脑部并移除脑膜。将脑放置在冰冷的完全dmem/f12培养中并保持在冰上直到处理。
[0340]
脑组织处理
[0341]
将鼠脑从培养基通过40μm细胞过滤器过滤移出。将脑转置于温的(37℃)0.25%胰蛋白酶(trypsin)(2ml/脑)并于37℃旋转下培养30分钟。以等体积的完全dmem/f12中止解离反应。将组织以300xg离心7分钟。将组织团块以完全dmem/f12清洗3次,以300xg离心7分钟。最终的清洗后,将组织团块再悬浮于完全dmem/f12中(~1ml/脑),研碎直到看不见大团块,然后经由70μm细胞过滤器过滤。将细胞分布于t150组织培养烧瓶(1支烧瓶/小鼠),每支烧瓶含至高最终体积35ml的完全dmem/f12。将细胞于第5天开始以每3至4天更换一次完全培养基来喂养直到进行分离。。
[0342]
微胶细胞分离
[0343]
培养11至14天后,以pbs清洗细胞,及以5ml 0.25%胰蛋白酶处理并于10ml完全dmem/f12中研碎。将单一细胞悬浮液经由70μm细胞过滤器过滤,以200xg离心6分钟,并以1x108个细胞/ml再悬浮于分离陪养基中。将至高4ml的细胞转置于14ml聚苯乙烯facs试管。于各样本中加入50μl/ml大鼠血清并将样本培养5分钟。通过将等份量的混合液a和b混合,制备选择性混合液(1ml样本混合25μl的混合液a和25μl混合液b),并培养5分钟。将以试剂盒提供的easysep(stemcell technologies,vancouver ca)选择性混合液(每ml细胞50μl)
加到facs试管,以移液吸管尖充分混合并培养5分钟。将以试剂盒提供的easysep rapidspheres涡旋30秒,每ml细胞加入80μl,以移液吸管尖混合,并于室温培养3分钟。将分离培养基加至样本中满足所指的体积(就<3ml的样本为5ml-就>或=3ml的样本为10ml)。将试管置于easysep magnet中历时5分钟。以一流体运动,当试管仍在磁极中的同时将未标定的细胞倒出至缓冲液中并保持倒置2-3秒。从磁极移出试管,并加入适量的分离培养基,另再放置5分钟。进行磁性分离/清洗程序4分钟用以移除所有未标定的细胞。最后的冲洗后,将标定的cd11b 微胶细胞再悬浮于无血清nbactiv1培养基中,计数及以所欲的浓度再悬浮。
[0344]
体外表示法
[0345]
分离后,将微胶细胞以每孔1百万个细胞植入12孔盘并静息24小时。将微胶细胞以2.5μm的btk抑制剂预处理30分钟,及在预处理后加入聚集/复合的igg(50μg/ml)用以刺激细胞群,并将细胞培养6h或24h。以200μl qiazol解离细胞并送出进行rna分离和定序(genewiz,south plainfield nj)。
[0346]
依照标准rnaseq法在omicsoft’s array studio平台内进行rnaseq库数据的分析。一般而言,定序读数经过质量检查并使用ensembl.r89基因模型将样本映像至小鼠mm10参照序列。收集表达的测量值(转录)为fpkm和计数。每组4x生物性复制(群)应用一低数过滤(low count filter),其中仅保留在至少二次转录中具有转录数>15的群组。将低数过滤应用于fpkm计数数据。此外,仅保留映像至编码基因的转录。在应用这些过滤后,保留12,790fpkm转录。将该fpkm数据分位数标准化(quantile normalized)至第75个百分位数。在log2转换前,将数值加到所有的样本。
[0347]
就下列分别在6小时和24小时仅以igg处理及以igg和btk抑制剂处理之间的比较进行t试验。在所有的比较中,测定符合下列标准的转录的差异表达基因(deg):倍数变化>1.2或<-1.2&p-值<0.05(fc1.2,p0.05)。
[0348]
通过辨识以活化状态增加的deg建立6h btki基因标记,其在以btk抑制剂处理后校正。通过辨识以活化状态增加的deg建立24h btki基因标记,其为在以btk抑制剂处理后校正,如图4所示。
[0349]
24h的rgs1 mrna的fpkm比较如图5a所示。
[0350]
体内初次受试小鼠(mouse)微胶细胞的处理
[0351]
如上述从产后小鼠制备小鼠初代微胶细胞,并以2.5μm的btki抑制剂处理6和24小时。分离rna并以定量实时pcr(taqman,thermo fisher)分析。相较于对照组的6h相对表达量如图5b所示(t-试验,*p<0.05)。
[0352]
体内小鼠研究
[0353]
每日以btk抑制剂或媒剂(100%peg200)的口服溶液治疗初次受试c57b16小鼠历时5天。于第5天最后给剂后1h将小鼠安乐死并灌注pbs,之后将脑分离出。使用rneasy lipid tissue rna分离试剂盒(qiagen)从鼠脑分离rna。使用实时pcr和taqman探针定量rgs1(图6;单边anova,**p<0.01)。
[0354]
人脑单细胞核rnaseq库制备
[0355]
病患样本和处理
[0356]
冷冻保存的死后脑检体得自ucla脑银行或克里夫兰医疗中心快速尸检项目
(cleveland clinic rapid autopsy program)。组织检体主要由具有小部分周围灰质的皮质下白质所组成。细胞核悬浮液依照前述方法做修改从均质化的组织所制备。简言之,将大约200至400mg的组织置于冰上,以240mm蔗糖、24mm kcl、4.8mm mgcl2、9.59mm tris(ph 8.0)、0.97um dtt、0.1%(v/v)triton x-100、1x蛋白酶抑制剂(fisher,pi78429)、0.4单位/μl rnasein(fisher,pr-n2511)和0.2units/μl superasin(fisher,am2694)溶于无核酸酶的水中所组成的细胞核分离缓冲液处理。先将样本置于含有缓冲液的培养皿中并使用剃刀剁碎。然后将组织悬浮液使用玻璃的杜恩斯均质机均质并将均质液经由100微米细胞过滤器过滤。从均质液通过1-2回合的清洗(nuclei pure store缓冲液,sigma,s9183)和离心(4级,700g历时3分钟),制备粗细胞核悬浮液。然后将细胞核以核酸结合染剂(dapi,1∶500)标定,以细胞核分离缓冲液再清洗一次,并以离心形成团块(4级,700g,2分钟)。通过将粗团块再悬浮于细胞核分离缓冲液制备最终的细胞核悬浮液并使用bd influx细胞分选仪将dapi 者分选于收集缓冲液中(0.5%ultrapure bsa和1x superasin溶于pbs中)。
[0357]
单细胞核rnaseq库制备
[0358]
依制造商说明(10x genomics)以v2 chemistry以chromium controller使用铬单细胞3’库试剂盒(chromium single cell 3’library kit)将分离的细胞核进行snrna-seq。使用nextseq500(illumina)将细胞核库定序。
[0359]
单细胞核rnaseq数据处理
[0360]
使用cell ranger analysis pipeline(10x genomics)进行样本多路分解(demultiplexing),条形码处理和单细胞计数。以说明外显子和内含子二者之前-mrna批注将rnaseq读数映射到人类参照基因组。使用r/bioconductor和partek single cell toolkit(partek)进行后续的数据分析。就质性对照,依照细胞将具有高粒线体含量、高umi和高基因数的细胞核移出。使用1,000,000的倍率将数据正常化。
[0361]
结果
[0362]
如图4中所示,微胶细胞的基因表达标记被igg活化改变,且此举被btk抑制作用正常化了。如图4中所示,rgs1经辨识为此btk-依赖的微胶细胞标记中上调的其中一个基因。rgs1(g蛋白信号传递1的调节子)作为一g蛋白信号除传递路径的负调节子且已牵涉各种发炎疾病。rgs1经辨识为一ms危险因子且亦发现富含在微胶细胞中(international multiple sclerosis genetics consortium,science 365:6460(2019))。
[0363]
当将体内仅以igg治疗及以igg和btk抑制剂治疗后的小鼠微胶细胞中的mrna表达相比较时,如图5a中所示,经igg治疗的rgs1的mrna表达上调并被btk抑制剂阻断。如图5b中所示,当以btk抑制剂处理初次受试小鼠的微胶细胞时,rgs1 mrna量下降。
[0364]
活体中btk抑制剂亦降低了rgs1表达。当每天一次以btk抑制剂治疗初次受试小鼠历时5天时,如图6中所示,在各种剂量(0.6、6和24mg/kg)观察到rgs1 mrna表达下降。这些结果指出,在体外和体内,btk调节了构成性rgs1脑表达,其可能预示了btk活性。
[0365]
图7a显示,使用单细胞rna seq数据集,从续发进展性ms(spms)微胶细胞辨识各种cns细胞,包括微胶细胞(以空心圆表示)。当检验单细胞mrna定序数据集的特定基因时,rgs1是spms微胶细胞中其中一种最上调的基因(表31和图7b)。
[0366]
表31
[0367]
rgs1上调显示在这些微胶细胞中btk可能是活化的,且rgs1亦可能具有与进展性ms微胶细胞表型相关联的独特功能。实施例4
[0368]
研究食物与btk抑制剂一起摄入的效应。进行实施例1的临床研究,并分析结果用以比较在禁食状态下以btk抑制剂治疗的病患和进食状态下治疗的病患。如上文实施例1.1中所述,从二组取得样本并分析用以评估药物动力学参数。
[0369]
结果确认,当相较于禁食状态下的给药时,在进食状态下施用btk抑制剂实质上增加中位数auc(曲线下面积)。具有较高auc的病患(较高的btk抑制剂暴露)产生非常少或无新的gd-增强的t1-高信号病灶。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。