一种合成艾曲波帕中间体3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的方法
技术领域
1.本发明属于有机药物合成技术领域,具体涉及一种艾曲波帕关键中间体3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的合成方法。
背景技术:
2.艾曲波帕(eltrombopag)是由英国葛兰素史克公司研发,并于2008年11月获得fda批准上市,用于治疗经糖皮质激素类药物、免疫球蛋白治疗无效或脾切除术后慢性特发性血小板减少性紫癜患者的血小板减少。随后,分别于2012年和2014年fda批准艾曲波帕用于慢性丙肝血小板减少患者和治疗对免疫疗法没有充分响应的严重型再生障碍性贫血患者,进一步扩大了其适应症。
[0003]3’‑
氨基-2
’‑
羟基联苯-3-羧酸(1)是制备艾曲波帕的关键中间体,其与1-(3,4-二甲苯基)-3-甲基-1h-吡唑-5(4h)-酮(2)反应后,再与乙醇胺(3)成盐可制得艾曲波帕api。
[0004][0005]
目前,3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的合成工艺主要有2种策路。
[0006]
第一种策略是以2-溴-6-硝基苯酚(4)为原料,选用合适的保护基保护羟基,再与3-羧基苯硼酸发生偶联反应得到联苯中间体,最后经过脱保护、还原硝基等步骤得到3
’‑
氨基-2
’‑
羟基联苯-3-羧酸。路线1采用碘甲烷甲基化保护羟基得到中间体5,再经过偶联、脱甲基保护、还原硝基得到目标产物。路线1采用剧毒且不易购买的碘甲烷,同时还需要使用强腐蚀性的溴化氢溶液,污染大,而且偶联反应需要在105度高温长时间反应43小时,难以实现大规模生产。路线2将路线1中甲基保护换成苄基保护,再发生偶联反应得到中间体6,最后经钯碳催化加氢脱除苄基同时还原硝基得到目标产物。路线2中溴化苄成本高且脱苄基需要高压加氢操作,具有一定的危险性。路线3先还原硝基为氨基得到中间体7,再采用n,n'-羰基二咪唑发生成环反应同时保护氨基和羟基得到中间体8,然后经偶联反应和水解脱保护制备得到目标产品。这一路线采用剧毒且不易存储和运输的n,n'-羰基二咪唑,而且偶联反应产率较低,因此难以实现大规模生产。
[0007][0008]
第二种策略是以对氯苯酚(10)为原料,通过在酚羟基对位引入氯原子占位,使苯环上反应只能发生于羟基的邻位,有效控制硝化反应和溴代反应过程中异构体杂质的生成,最后氯原子在硝基还原反应中同时被去除掉,制备3
’‑
氨基-2
’‑
羟基联苯-3-羧酸。路线4和路线5中,分别针对硝化反应、溴代反应和偶联反应采取了不同的反应顺序,都制备了目标产物,但是由于氯原子引入导致偶联反应的收率较差,生产成本较高,同时需使用高沸点的聚乙二醇为溶剂,后处理繁琐,去除及回收困难,增加了三废处理难度,并不适用于大规模的工业化生产。
[0009][0010]
现有技术的诸多不足制约了3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的大规模工业化生产及成本降低,因此有必要开发容易控制、安全环保、操作简单、产率高,适合工业生产的合成方法。
技术实现要素:
[0011]
针对以上弊端,本发明公开了一种制备3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的新方法,该方法所用原料易得,操作步骤简便,成本低、收率高,经济环保,有利于实现工业化生产。
[0012]
本发明的技术解决方案包括如下步骤:一种合成3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的方法,包括:
[0013]
(1)以2-溴-6-硝基苯酚4为原料经过硝基还原反应制备2-氨基-6-溴苯酚7的步骤,
[0014]
[0015]
(2)将2-氨基-6-溴苯酚7和r2cho在催化剂作用下经过环化反应得到化合物17的步骤,
[0016][0017]
r2为苯基、对甲基苯基、对氯苯基、对硝苯基、1-萘基中的任意一种;
[0018]
(3)将化合物17和3-羧基苯硼酸经suzuki偶联反应得制得化合物18的步骤,
[0019][0020]
(4)将化合物18经水解反应得目标产物3
’‑
氨基-2
’‑
羟基联苯-3-羧酸1的步骤,
[0021][0022]
较佳的,步骤(1)中,硝基还原反应的还原剂采用氢源,氢源选自甲酸铵、水合肼中的任一种;反应体系采用有机溶剂作为溶剂,有机溶剂选自甲醇、乙醇、丙醇、乙腈、丙酮、四氢呋喃、1,4-二氧六环、甲基叔丁基醚、乙酸乙酯、n,n-二甲基甲酰胺、二甲基亚砜中的任一种;催化剂选自钯碳(钯含量1%-10wt%)、铂碳(铂含量1%-10wt%)、雷尼镍中的任一种。
[0023]
具体的,2-溴-6-硝基苯酚与催化剂的质量比为1:0.01~1;2-溴-6-硝基苯酚与氢源的摩尔比为1:2~10;反应温度为30~80℃;反应时间为3~10h。
[0024]
较佳的,步骤(2)中,催化剂采用杂多酸类离子液体催化剂,杂多酸类离子液体催化剂选自[mimps]3pw
12o40
、[mimps]3pmo
12o40
、[pyps]3pw
12o40
、[pyps]3pmo
12o40
、[teaps]3pw
12o40
、[teaps]3pmo
12o40
中的任意一种,其化学结构为:
[0025][0026]
较佳的,步骤(2)中,2-氨基-6-溴苯酚、r2cho、催化剂的摩尔比为1:1~2:0.01~0.05;反应温度为60~120℃;反应时间为6~24h。
[0027]
较佳的,步骤(3)中,反应在碱和催化剂存在下进行,催化剂选自钯碳(钯含量1%-10wt%)、pdcl2(pph3)2、pd(pph3)4、pd(oac)2、pdcl2中的任意一种;碱选自碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、碳酸铯、氢氧化钠和氢氧化钾中的任意一种;反应体系采用有机溶剂和水的混合物作为溶剂,有机溶剂选自甲醇、乙醇、丙醇、四氢呋喃、1,4-二氧六环、甲基叔
丁基醚、n,n-二甲基甲酰胺、二甲基亚砜中的任意一种。
[0028]
具体的,化合物17、3-羧基苯硼酸、碱、催化剂的摩尔比为1:1~2:1.5~2.5:0.01~0.1;有机溶剂和水的体积比为5~1:1;反应温度为50~100℃;反应时间为3~12h。
[0029]
较佳的,步骤(4)中,反应体系在碱存在下进行,碱选自氢氧化钾、氢氧化钠、氢氧化钙、氢氧化钡、碳酸钾和碳酸铯中的任意一种。
[0030]
具体的,化合物18和碱的摩尔比为1:10~30;反应温度为60~110℃;反应时间为4~24h。
[0031]
与现有技术相比,本发明的有益效果是:(1)硝基还原步骤原料廉价易得,使用的氢源操作便捷、安全;环化保护步骤使用杂多酸类离子液体催化剂合成方法简易、高活性、可多次回收套用、绿色环保;suzuki偶联步骤采用价格低、来源广泛易得的钯碳为催化剂,简单过滤分离后可以直接回收套用;水解脱保护步骤操作简单、转化率高。(2)整个过程避免使用诸剧毒化学品,工艺操作简便,条件易控制,试剂和中间体稳定性高,便于运输和储存,安全隐患较小,废液处理容易,绿色环保,便于工业化大规模使用。
附图说明
[0032]
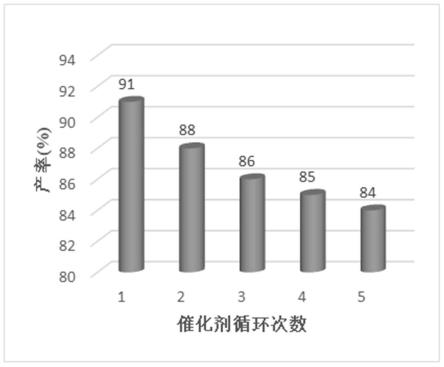
图1为实施例4中b组为模板反应催化剂杂多酸类离子液体的活性重复性实验效果图。
具体实施方式
[0033]
以下通过实施例进一步说明本发明,但专利权利并不局限于这些实施例。
[0034]
本发明的所述的艾曲波帕关键中间体3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的合成方法,采用2-溴-6-硝基苯酚为原料,经过硝基还原、环化保护、suzuki偶联和水解脱保护四步反应得到目标产物,具体包括:
[0035]
步骤1:以2-溴-6-硝基苯酚(4)为原料经过硝基还原反应制备2-氨基-6-溴苯酚(7),
[0036][0037]
搅拌条件下,向有机溶剂中依次加入2-溴-6-硝基苯酚(4)和催化剂,再缓慢加入氢源,升温达到目标反应温度后保温反应一定时间,待反应完毕后降温至室温,过滤回收催化剂可套用,浓缩滤液后可进一步通过乙醇重结晶或柱层析纯化得到2-氨基-6-溴苯酚(7)。
[0038]
步骤2:2-氨基-6-溴苯酚(7)和r2cho经过环化反应得到化合物17,
[0039]
[0040]
搅拌条件下,向反应器中依次加入2-氨基-6-溴苯酚(7)、r2cho和杂多酸类离子液体催化剂,升温达到目标反应温度后保温反应一定时间,待反应完毕后降温至室温,向反应器内加入非质子性弱极性溶剂(非质子性弱极性溶剂为乙酸乙酯、二氯甲烷中的任一种),搅拌溶解产物后,过滤回收杂多酸类离子液体催化剂,可循环重复使用,浓缩滤液可进一步通过乙醇重结晶或柱层析纯化得到化合物17。
[0041]
步骤3:化合物17和3-羧基苯硼酸经suzuki偶联反应得制得化合物18,
[0042][0043]
搅拌条件下,向有机溶剂和水的混合液中依次加入化合物17、3-羧基苯硼酸、碱和催化剂,氮气保护下升温达到目标反应温度后保温反应一定时间,待反应完毕后趁热过滤除去不溶物,滤液加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到化合物18。
[0044]
步骤4:化合物18经水解反应得3
’‑
氨基-2
’‑
羟基联苯-3-羧酸(1),
[0045][0046]
搅拌条件下,向水中依次加入化合物18和碱,升温达到目标反应温度后保温反应一定时间,待反应完毕后降温至室温,加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到3
’‑
氨基-2
’‑
羟基联苯-3-羧酸(1)。
[0047]
本发明所述的3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的合成方法,其合成路线如下:
[0048][0049]
实施例中所使用的杂多酸类离子液体催化剂的制备方法,参见相关文献(an eco-benign and highly efficient procedure for n-acylation catalyzed by heteropolyanion-based ionic liquids using carboxylic acid under solvent-free conditions,tetrahedron 70(2014)2237-2245.)。
[0050]
实施例1:2-氨基-6-溴苯酚的制备
[0051]
搅拌条件下,向甲醇150ml中依次加入2-溴-6-硝基苯酚10.0g和10wt%钯碳1.0g,再缓慢加入甲酸铵11.6g,升温达到64℃后保温反应6h,待反应完毕后降温至室温,过滤回收催化剂可套用,浓缩滤液后可进一步通过乙醇重结晶或柱层析纯化得到2-氨基-6-溴苯酚8.3g,产率96%。1h nmr(400m hz,cdcl3)δ8.28(brs,1h),6.89(d,j=8.0hz,1h),6.62-6.45(m,2h),5.33(brs,2h);
13
c nmr(100m hz,cdcl3)δ148.7,141.5,125.6,122.9,115.7,
144.8。
[0052]
实施例2:2-氨基-6-溴苯酚的制备
[0053]
搅拌条件下,向乙酸乙酯150ml中依次加入2-溴-6-硝基苯酚10.0g和10wt%铂碳1.0g,再缓慢加入水合肼11.5g,升温达到77℃后保温反应4h,待反应完毕后降温至室温,过滤回收催化剂可套用,浓缩滤液后可进一步通过乙醇重结晶或柱层析纯化得到2-氨基-6-溴苯酚8.2g,产率95%。1h nmr(400m hz,cdcl3)δ8.28(brs,1h),6.89(d,j=8.0hz,1h),6.62-6.45(m,2h),5.33(brs,2h);
13
c nmr(100m hz,cdcl3)δ148.7,141.5,125.6,122.9,115.7,144.8。
[0054]
实施例3:2-氨基-6-溴苯酚的制备
[0055]
搅拌条件下,向1,4-二氧六环250ml中依次加入2-溴-6-硝基苯酚20.0g和10wt%钯碳1.0g,再缓慢加入甲酸铵17.4g,升温达到100℃后保温反应8h,待反应完毕后降温至室温,过滤回收催化剂可套用,浓缩滤液后可进一步通过乙醇重结晶或柱层析纯化得到2-氨基-6-溴苯酚16.0g,产率93%。1h nmr(400m hz,cdcl3)δ8.28(brs,1h),6.89(d,j=8.0hz,1h),6.62-6.45(m,2h),5.33(brs,2h);
13
c nmr(100m hz,cdcl3)δ148.7,141.5,125.6,122.9,115.7,144.8。
[0056]
实施例4:化合物17a的制备
[0057][0058]
实验分为七组a-g组对比实验,每组加入不同种类的杂多酸类离子液催化剂或者不加催化剂
[0059]
a组:[mimps]3pmo
12o40
[0060]
b组:[mimps]3pw
12o40
[0061]
c组:[pyps]3pmo
12o40
[0062]
d组:[pyps]3pw12o
40
[0063]
e组:[teaps]3pmo
12o40
[0064]
f组:[teaps]3pw
12o40
[0065]
g组:不加催化剂
[0066]
具体实验方法如下:
[0067]
搅拌条件下,向反应器中依次加入2-氨基-6-溴苯酚3.0g、苯甲醛2.0g和分别a-g组催化剂0.16mmol,升温达到100℃后保温反应10h,待反应完毕后降温至室温,向反应器内加入乙酸乙酯60ml,搅拌溶解产物后,过滤回收杂多酸类离子液体催化剂,可循环重复使用,浓缩滤液可进一步通过乙醇重结晶或柱层析纯化得到化合物17a。1h nmr(400m hz,cdcl3)δ8.25(d,j=8.0hz,2h),7.72-7.68(m,3h),7.62-7.51(m,2h),7.19(t,j=7.8hz,1h);
13
c nmr(100m hz,cdcl3)δ168.5,151.2,145.1,132.9,129.9,128.3,127.8,127.1,126.5,120.1,109.2。
[0068]
表1 a-g组对比实验结果
[0069]
组别杂多酸类离子液体催化剂产率a组[mimps]3pmo
12o40
83%b组[mimps]3pw
12o40
91%c组[pyps]3pmo
12o40
80%d组[pyps]3pw
12o40
89%e组[teaps]3pmo
12o40
62%f组[teaps]3pw
12o40
75%g组不加催化剂21%
[0070]
由表1的数据可以得出以下结论:加入杂多酸类离子液体催化剂(a-f组)的实验结果明显优于不加催化剂(g组)的实验结果;含有mimps和pyps的杂多酸类离子液体的催化活性相当,且都高于含有teaps的杂多酸类离子液体;含有pw
12o40
的杂多酸类离子液体的催化活性高于含有pmo
12o40
的杂多酸类离子液体;a-f组中催化活性最高的是[mimps]3pw
12o40
(b组)。
[0071]
实施例5:
[0072]
以实施例4中b组为模板反应,进行催化剂杂多酸类离子液体的活性重复性试验,离子液体重复使用5次。反应的产率数据见图1。
[0073]
由图1和数据可以得出这样的结论:杂多酸类离子液体催化剂在循环使用过程中产率稍有降低,但降低幅度较小,因此,可以证明该杂多酸类离子液体可以循环使用。
[0074]
实施例6:化合物17b的制备
[0075][0076]
搅拌条件下,向反应器中依次加入2-氨基-6-溴苯酚3.0g、4-甲基苯甲醛2.3g和[mimps]3pw
12o40 1.1g,升温达到90℃后保温反应8h,待反应完毕后降温至室温,向反应器内加入乙酸乙酯80ml,搅拌溶解产物后,过滤回收杂多酸类离子液体催化剂,可循环重复使用,浓缩滤液可进一步通过乙醇重结晶或柱层析纯化得到化合物17b 4.24g,产率92%。1h nmr(400m hz,cdcl3)δ8.02(d,j=8.0hz,2h),7.59-7.51(m,2h),7.32-7.21(m,3h),2.44(s,3h);
13
c nmr(100m hz,cdcl3)δ167.2,151.8,144.2,130.2,128.8,128.0,127.3,126.8,126.0,119.5,108.6,24.2。
[0077]
实施例7:化合物17c的制备
[0078][0079]
搅拌条件下,向反应器中依次加入2-氨基-6-溴苯酚3.0g、4-氯苯甲醛2.7g和[pyps]3pmo
12o40 0.77g,升温达到100℃后保温反应12h,待反应完毕后降温至室温,向反应器内加入乙酸乙酯80ml,搅拌溶解产物后,过滤回收杂多酸类离子液体催化剂,可循环重复
使用,浓缩滤液可进一步通过乙醇重结晶或柱层析纯化得到化合物17c 4.34g,产率88%。1h nmr(400m hz,cdcl3)δ7.74(d,j=8.0hz,2h),7.52-7.47(m,4h),7.27-7.24(m,1h);
13
c nmr(100m hz,cdcl3)δ165.5,154.8,143.1,135.1,129.3,127.8,126.6,125.8,125.0,118.5,107.7。
[0080]
实施例8:化合物18a的制备
[0081][0082]
搅拌条件下,向1,4-二氧六环250ml和水50ml的混合液中依次加入化合物17a 12.3g、3-羧基苯硼酸9.0g、碳酸钾12.4g和10%钯碳2.0g,氮气保护下升温达到100℃后保温反应4h,待反应完毕后趁热过滤除去不溶物,滤液加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到化合物18a 12.7g,产率90%。1h nmr(400m hz,dmso-d6)δ8.75(s,1h),8.26-8.25(m,1h),8.24-8.12(m,2h),7.88-7.85(m,1h),7.79-7.76(m,3h),7.67-7.62(m,3h),7.57-7.54(m,1h);
13
c nmr(100m hz,dmso-d6)δ170.1,167.2,149.8,143.1,137.5,134.1,131.0,130.7,129.4,129.2,129.0,128.0,127.2,126.5,125.9,125.1,119.1,118.3。
[0083]
实施例9:化合物18b的制备
[0084][0085]
搅拌条件下,向甲醇200ml和水100ml的混合液中依次加入化合物17b 13.0g、3-羧基苯硼酸8.2g、碳酸钠9.5g和pd(pph3)4 2.0g,氮气保护下升温达到80℃后保温反应6h,待反应完毕后趁热过滤除去不溶物,滤液加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到化合物18b 12.1g,产率82%。1h nmr(400m hz,dmso-d6)δ8.71(s,1h),8.25-8.24(m,1h),7.98-7.92(m,2h),7.85-7.81(m,1h),7.75-7.69(m,3h),7.54-7.50(m,1h),7.27-7.26(m,2h),2.30(s,3h);
13
c nmr(100m hz,dmso-d6)δ169.8,165.8,148.1,142.1,136.0,133.1,130.4,130.1,129.0,128.8,127.7,127.2,126.2,125.8,125.0,125.1,118.4,117.9,23.5。
[0086]
实施例10:化合物18c的制备
[0087][0088]
搅拌条件下,向n,n-二甲基甲酰胺150ml和水150ml的混合液中依次加入化合物17c 13.9g、3-羧基苯硼酸9.0g、碳酸铯26.4g和pd(oac)
2 2.0g,氮气保护下升温达到100℃后保温反应3h,待反应完毕后趁热过滤除去不溶物,滤液加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到化合物18c 13.4g,产率85%。1h nmr(400m hz,dmso-d6)δ8.73(s,1h),8.23-8.21(m,1h),7.79-7.77(m,2h),7.76-7.81(m,1h),7.75-7.69(m,3h),7.53-7.49(m,3h);
13
c nmr(100m hz,dmso-d6)δ168.7,164.7,147.7,142.0,136.1,134.1,133.4,130.2,134.1,128.9,128.6,128.0,127.1,126.5,125.9,125.2,118.0,117.5。
[0089]
实施例11:3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的制备
[0090]
搅拌条件下,向水120ml中依次加入化合物18a 9.5g和氢氧化钾30.0g,升温达到100℃后保温反应8h,待反应完毕后降温至室温,加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到3
’‑
氨基-2
’‑
羟基联苯-3-羧酸(1)6.4g,产率93%。1h nmr(400mhz,dmso-d6)δ8.07(s,1h),7.88(d,j=7.7hz,1h),7.70(d,j=7.8hz,1h),7.52(t,j=7.7hz,1h),6.68(t,j=7.8hz,1h),6.64(dd,j=7.8,1.1hz,1h),6.47(dd,j=7.2,1.2hz,1h)
[0091]
实施例12:3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的制备
[0092]
搅拌条件下,向水120ml中依次加入化合物18b 9.9g和氢氧化钠26.4g,升温达到100℃后保温反应10h,待反应完毕后降温至室温,加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到3
’‑
氨基-2
’‑
羟基联苯-3-羧酸(1)6.2g,产率90%。1h nmr(400mhz,dmso-d6)δ8.07(s,1h),7.88(d,j=7.7hz,1h),7.70(d,j=7.8hz,1h),7.52(t,j=7.7hz,1h),6.68(t,j=7.8hz,1h),6.64(dd,j=7.8,1.1hz,1h),6.47(dd,j=7.2,1.2hz,1h)
[0093]
实施例13:3
’‑
氨基-2
’‑
羟基联苯-3-羧酸的制备
[0094]
搅拌条件下,向水120ml中依次加入化合物18c 10.5g和氢氧化钾33.6g,升温达到80℃后保温反应10h,待反应完毕后降温至室温,加入稀盐酸调节ph至酸性,过滤得滤饼可进一步通过乙醇重结晶或柱层析纯化得到3
’‑
氨基-2
’‑
羟基联苯-3-羧酸6.3g,产率91%(1)。1h nmr(400mhz,dmso-d6)δ8.07(s,1h),7.88(d,j=7.7hz,1h),7.70(d,j=7.8hz,1h),7.52(t,j=7.7hz,1h),6.68(t,j=7.8hz,1h),6.64(dd,j=7.8,1.1hz,1h),6.47(dd,j=7.2,1.2hz,1h)。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
