1.本发明涉及有机电致发光材料技术领域,具体涉及一种基于聚氨酯主链的聚合物发光材料及其制备方法和应用。
背景技术:
2.由于有机发光二极管(oled)在照明、可穿戴、柔性显示和大尺寸显示中具有传统液晶显示(lcd)所不能比拟的优点,使其受到了越来越广泛的应用。其中基于旋涂和喷墨打印等湿法工艺制备的oled器件在柔性显示和大尺寸显示方面具有真空蒸镀法所不能比拟的优势,十分利于工业化大规模生产。其中,聚合物发光单元是湿法器件当中最为重要的一环,制备高性能的聚合物发光二极管(pled)是目前的研究热点。
3.现有的聚合物发光材料根据发光机理可以分为以下三种:荧光聚合物、磷光聚合物和热活化延迟荧光(tadf)聚合物分子。tadf起源于2012年,日本九州大学chihaya adachi教授在《自然》杂志上发表了题为“来源于延迟荧光的高效有机发光二极管”(uoyama,h.,goushi,k.,shizu,k.et al.nature 492,234-238(2012).)的论文,阐释了tadf的理念,将荧光材料的理论内量子效率提高到100%,具有很大的商业潜力,被视为下一代oled显示的关键技术。相较于传统的荧光或磷光聚合物发光材料,tadf聚合物发光材料具有高激子利用率和高效率,因此受到了越来越广泛的关注。
4.目前基于tadf的聚合物材料的主要结构是以共轭单元为主链,侧链为tadf给受体结构(yun yang,lei zhao,shumeng wang,junqiao ding,and lixiang wang et al.macromolecules 2018 51(23),9933-9942),这样的结构便于调整给受体之间的组合,从而可以实现发光颜色的调控。但是这样的共轭主链在聚集态下的相互作用很强,会使得聚合物在薄膜状态下的光致发光量子产率(plqy)急剧下降,形成聚集猝灭效应。并且这样的共轭主链成膜性欠佳,难以达到很高的聚合度,限制了其在电致发光器件上的应用。
技术实现要素:
5.本发明的目的在于提供一种基于聚氨酯主链的聚合物发光材料及其制备方法和应用,本发明提供的聚合物发光材料能够提高光致发光量子产率,并且具有更好的溶解性和成膜性,有助于提高其在电致发光器件中的性能。
6.为了实现上述发明目的,本发明提供以下技术方案:
7.本发明提供了一种基于聚氨酯主链的聚合物发光材料,具有式i所示结构:
8.9.式i中,r1为喹喔啉基或2,3-二氰基喹喔啉基;r2为咔唑基、三苯胺基或9,9-二甲基-9,10二氢吖啶基;x z的范围为0.05~0.5,y的范围为0.5~0.95。
10.优选地,所述基于聚氨酯主链的聚合物发光材料包括
[0011][0012]
本发明提供了上述技术方案所述聚合物发光材料的制备方法,包括以下步骤:
[0013]
当r2为咔唑基或三苯胺基时,在保护气氛下,将x-r
1-x、碱性试剂i、钯催化剂i和溶剂i混合,进行第一偶联反应,得到r
2-r
1-x;
[0014]
当r2为9,9-二甲基-9,10二氢吖啶基时,在保护气氛下,将x-r
1-x、9,9-二甲基-9,10二氢吖啶、碱性试剂ii、钯催化剂ii、催化剂配体和溶剂ii混合,进行c-n偶联反应,得到r
2-r
1-x;
[0015]
所述x-r
1-x和r
2-r
1-x中x为溴基或碘基;
[0016]
将所述r
2-r
1-x和3,5-二羟基苯硼酸、碱性试剂iii、钯催化剂iii以及溶剂iii混合,进行第二偶联反应,得到具有式ii所示结构的化合物;
[0017]
将所述具有式ii所示结构的化合物和1,6-己二异氰酸酯以及2,2-二羟甲基丙酸混合,进行扩链反应,得到具有式i所示结构的聚合物发光材料;
[0018][0019]
优选地,所述溶剂i和溶剂iii独立为甲苯和水。
[0020]
优选地,所述第一偶联反应的温度为80~110℃;所述第一偶联反应的保温时间为10~14h。
[0021]
优选地,所述溶剂ii为甲苯。
[0022]
优选地,所述第二偶联反应的温度为80~110℃;所述第二偶联反应的保温时间为
10~14h。
[0023]
优选地,所述保护气氛为氮气气氛。
[0024]
优选地,所述扩链反应的温度为85~90℃。
[0025]
本发明提供了上述技术方案所述聚合物发光材料或上述技术方案所述制备方法制备得到的聚合物发光材料作为有机电致发光材料的应用。
[0026]
本发明提供了一种基于聚氨酯主链的聚合物发光材料,本发明采用聚氨酯型的非共轭主链可以减小聚集态下的相互作用,有效地降低因为聚合物分子长链的π-π*堆积所导致的聚集猝灭效应,提高光致发光量子产率;并且聚氨酯型主链可以很方便的调节软硬链段之间的比例,从而可以显著改善聚合物的溶解性和成膜性,有助于提高其在电致发光器件中的性能。本发明采用聚氨酯主链有利于实现聚合,并且能够达到很高的聚合度,将tadf给受体单元作为侧链引入聚合物,可以实现发光颜色的调控。
附图说明
[0027]
图1为实施例1制备的聚合物发光材料在甲苯中的荧光光谱图;
[0028]
图2为实施例2制备的聚合物发光材料在甲苯中的荧光光谱图;
[0029]
图3为实施例3制备的聚合物发光材料在甲苯中的荧光光谱图。
具体实施方式
[0030]
本发明提供了一种基于聚氨酯主链的聚合物发光材料,具有式i所示结构:
[0031][0032]
式i中,r1为喹喔啉基或2,3-二氰基喹喔啉基;r2为咔唑基、三苯胺基或9,9-二甲基-9,10二氢吖啶基;x z的范围为0.05~0.5,y的范围为0.5~0.95。
[0033]
在本发明中,当r1为喹喔啉基时,r2为咔唑基、三苯胺基或9,9-二甲基-9,10二氢吖啶基;当r1为2,3-二氰基喹喔啉基时,r2为咔唑基、三苯胺基或9,9-二甲基-9,10二氢吖啶基。
[0034]
在本发明中,所述喹喔啉基的结构为所述2,3-二氰基喹喔啉基的结构为所述咔唑基的结构为所述三苯胺基的结构为
所述9,9-二甲基-9,10二氢吖啶基的结构为
[0035]
在本发明中,x y z=1。在本发明的具体实施例中,x=0.1~0.2,z=0.4~0.3,y=0.5。
[0036]
在本发明中,所述基于聚氨酯主链的聚合物发光材料的数均分子量优选为4699~5497da;重均分子量优选为7330~8190da;分散度优选为1.49~1.56。
[0037]
在本发明中,所述基于聚氨酯主链的聚合物发光材料优选包括
[0038][0039]
本发明提供了上述技术方案所述聚合物发光材料的制备方法,包括以下步骤:
[0040]
当r2为咔唑基或三苯胺基时,在保护气氛下,将x-r
1-x、碱性试剂i、钯催化剂i和溶剂i混合,进行第一偶联反应,得到r
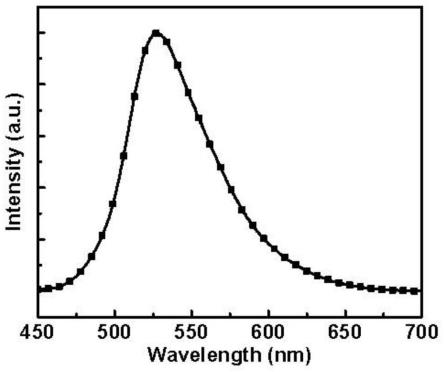
2-r
1-x;
[0041]
当r2为9,9-二甲基-9,10二氢吖啶基时,在保护气氛下,将x-r
1-x、9,9-二甲基-9,10二氢吖啶、碱性试剂ii、钯催化剂ii、催化剂配体和溶剂ii混合,进行c-n偶联反应,得到r
2-r
1-x;
[0042]
所述x-r
1-x和r
2-r
1-x中x为溴基或碘基;
[0043]
将所述r
2-r
1-x和3,5-二羟基苯硼酸、碱性试剂iii、钯催化剂iii以及溶剂iii混合,进行第二偶联反应,得到具有式ii所示结构的化合物;
[0044]
将所述具有式ii所示结构的化合物和1,6-己二异氰酸酯以及2,2-二羟甲基丙酸混合,进行扩链反应,得到具有式i所示结构的聚合物发光材料;
[0045]
[0046]
当r2为咔唑基或三苯胺基时,本发明在保护气氛下,将x-r
1-x、碱性试剂i、钯催化剂i和溶剂i混合,进行第一偶联反应,得到r
2-r
1-x。在本发明中,所述保护气氛优选为氮气气氛。在本发明中,所述x-r
1-x和r
2-r
1-x中的x为溴基或碘基,优选为br。
[0047]
在本发明中,所述x-r
1-x和的摩尔比优选为1~1.5:1,更优选为1.1~1.4:1。在本发明中,所述碱性试剂i优选包括碳酸钠、醋酸钠或碳酸钾;所述x-r
1-x和碱性试剂i的摩尔比优选为10~15:20~25。在本发明中,所述钯催化剂i优选包括四(三苯基膦)钯、醋酸钯或三(二亚苄基丙酮)二钯;所述x-r
1-x和钯催化剂i的摩尔比优选为10~15:0.5~0.75。在本发明中,所述溶剂i优选为甲苯和水;所述甲苯和水的体积比优选为40~60:10~15。在本发明中,所述x-r
1-x和溶剂i的用量比优选为10~15mmol:50~75ml。
[0048]
在本发明中,所述第一偶联反应的温度优选为80~110℃,更优选为90~100℃;所述第一偶联反应的保温时间优选为10~14h,更优选为12~13h。
[0049]
本发明在所述第一偶联反应后,优选还包括后处理;所述后处理优选包括依次进行的过滤、萃取、旋干和柱层析。在本发明中,所述萃取用试剂优选为乙酸乙酯或二氯甲烷;所述柱层析用试剂优选为正己烷、乙酸乙酯和二氯甲烷中的一种或几种。
[0050]
当r2为9,9-二甲基-9,10二氢吖啶基时,本发明在保护气氛下,将x-r
1-x、9,9-二甲基-9,10二氢吖啶、碱性试剂ii、钯催化剂ii、催化剂配体和溶剂ii混合,进行c-n偶联反应,得到r
2-r
1-x;所述x-r
1-x和r
2-r
1-x中x为溴基或碘基,优选为br。
[0051]
在本发明中,所述x-r
1-x和9,9-二甲基-9,10二氢吖啶的摩尔比优选为1~1.5:1,更优选为1.1~1.4:1。在本发明中,所述碱性试剂ii优选为叔丁醇钠;所述x-r
1-x和碱性试剂ii的摩尔比优选为10~15:20~25。在本发明中,所述钯催化剂ii优选包括四(三苯基膦)钯、醋酸钯或三(二亚苄基丙酮)二钯;所述x-r
1-x和钯催化剂ii的摩尔比优选为10~15:0.5~0.75。在本发明中,所述催化剂配体优选为(三叔丁基)膦四氟硼酸盐;所述钯催化剂ii和催化剂配体的摩尔比优选为0.5:1。在本发明中,所述溶剂ii优选为甲苯。在本发明中,所述x-r
1-x和溶剂ii的用量比优选为10~15mmol:40~75ml。
[0052]
在本发明中,所述c-n偶联反应的温度优选为80~110℃,更优选为90~100℃;所述c-n偶联反应的保温时间优选为10~14h,更优选为12~13h。
[0053]
本发明在所述c-n偶联反应后,优选还包括后处理;所述后处理优选包括依次进行的过滤、萃取、旋干和柱层析。在本发明中,所述萃取用试剂优选为乙酸乙酯或二氯甲烷;所述柱层析用试剂优选为正己烷、乙酸乙酯和二氯甲烷中的一种或几种。
[0054]
得到r
2-r
1-x后,本发明将所述r
2-r
1-x和3,5-二羟基苯硼酸、碱性试剂iii、钯催化剂iii以及溶剂iii混合,进行第二偶联反应,得到具有式ii所示结构的化合物。在本发明中,所述3,5-二羟基苯硼酸的结构式为在本发明中,所述r
2-r
1-x和3,5-二羟基苯硼酸的摩尔比优选为1:1~1.5。在本发明中,所述碱性试剂iii优选包括碳酸钠、醋酸
钠或碳酸钾;所述r
2-r
1-x和碱性试剂iii的摩尔比优选为10~15:20~25。在本发明中,所述钯催化剂iii优选包括四(三苯基膦)钯、醋酸钯或三(二亚苄基丙酮)二钯;所述r
2-r
1-x和钯催化剂iii的摩尔比优选为10~15:0.5~0.75。在本发明中,所述溶剂iii优选为甲苯和水;甲苯和水体积比优选为4~6:1;所述r
2-r
1-x和溶剂iii的用量比优选为10~15mmol:40~60ml。
[0055]
在本发明中,所述第二偶联反应优选在保护气氛中进行,更优选在氮气气氛中进行。在本发明中,所述第二偶联反应的温度优选为80~110℃,更优选为90~100℃;所述第二偶联反应的保温时间优选为10~14h,更优选为12~13h。
[0056]
本发明在所述第二偶联反应后,优选还包括后处理;所述后处理包括依次进行的过滤、萃取、旋干和柱层析。在本发明中,所述萃取用试剂优选为乙酸乙酯或二氯甲烷;所述柱层析用试剂优选为正己烷、乙酸乙酯和二氯甲烷中的一种或几种。
[0057]
在本发明中,所述具有式ii所示结构的化合物为
[0058]
得到具有式ii所示结构的化合物后,本发明将所述具有式ii所示结构的化合物和1,6-己二异氰酸酯以及2,2-二羟甲基丙酸混合,进行扩链反应,得到具有式i所示结构的聚合物发光材料。在本发明中,所述具有式ii所示结构的化合物、1,6-己二异氰酸酯和2,2-二羟甲基丙酸的配比以满足式i所示结构为准。在本发明中,所述混合优选包括:将所述具有式ii所示结构的化合物和1,6-己二异氰酸酯混合,升温至75~80℃搅拌2小时,然后加入2,2-二羟甲基丙酸混合。
[0059]
在本发明中,所述扩链反应的温度优选为85~90℃。本发明优选采用tlc监测反应进程,具体优选为:体系升温至85~90℃进行扩链反应,间隔2小时点板观察hdi浓度,当hdi浓度降到最低时,加入丙酮降低粘度并用三乙胺淬灭hdi。
[0060]
本发明优选在所述扩链反应后,还包括后处理;所述后处理包括:将得到的反应溶液倒入甲醇中析出沉淀,过滤得到固体;将固体加入盐酸溶液中搅拌溶解,然后以氯仿萃取,分离并收集有机相;向有机相中加入水,萃取分离,重复进行3次,收集剩余有机相;将有机相经真空旋转蒸发浓缩,浓缩液倒入甲醇中析出沉淀,过滤得到固体,然后将固体置于索氏提纯器中,以丙酮提纯后,干燥,得到具有式i所示结构的聚合物发光材料。在本发明的具体实施例中,将扩链反应得到的反应溶液倒入甲醇中搅拌25~30min析出沉淀,过滤得到固体;将固体加入2mol/l的盐酸溶液中搅拌溶解,然后以氯仿萃取,分离并收集有机相;向有机相中加入去离子水,搅拌5~25min,萃取分离,重复进行3次,收集剩余有机相;将有机相经真空旋转蒸发浓缩,浓缩液倒入甲醇中搅拌25~30min析出沉淀,过滤得到固体,然后将固体置于索氏提纯器中,以丙酮提纯24~48h后,于50~75℃真空干燥箱中干燥4~8h,得到具有式i所示结构的聚合物发光材料。
[0061]
在本发明中,所述具有式i所示结构的聚合物发光材料的得率优选为30~60%;纯度优选》99%。
[0062]
本发明还提供了上述技术方案所述聚合物发光材料或上述技术方案所述制备方法制备得到的聚合物发光材料作为有机电致发光材料的应用。以本发明提供的聚合物发光
材料制成结构为ito/聚3,4-乙撑二氧噻吩/聚苯乙烯磺酸盐(pedot:pss)/具有式i所示结构的聚合物发光材料/1,3,5-三(间吡啶-3-基苯基)苯(tmpypb)/lif/al的有机电致发光器件,电致发光颜色范围为黄光到红光,且电致发光光谱范围较宽。在本发明中,所述pedot:pss的厚度优选为40nm;所述具有式i所示结构的聚合物发光材料的厚度优选为40~50nm;所述tmpypb的厚度优选为40~50nm;所述lif的厚度优选为1nm;所述al的厚度优选为150nm。
[0063]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0064]
实施例1
[0065]
以三苯胺和喹喔啉作为给受体组合的聚合物发光材料的合成
[0066]
(1)4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺的合成
[0067][0068]
在干燥洁净的三口烧瓶中加入11mmol 4-硼酸三苯胺和10mmol 1,4-二溴喹喔啉,以及20mmol无水碳酸钠,并加入40ml无水甲苯和10ml水,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol四(三苯基膦)钯,随后继续抽真空通氮气三次,然后在100℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺。
[0069]1h nmr(500mhz,dmso-d6)δ8.76
–
8.68(m,1h),7.99
–
7.91(m,1h),7.66
–
7.60(m,1h),7.33
–
7.27(m,2h),7.27
–
7.22(m,1h),7.14
–
7.08(m,2h),7.07(tt,j=7.4,1.4hz,1h)。
[0070]
(2)(4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺基)-1,3-二羟基苯酚的合成
[0071][0072]
在干燥洁净的三口烧瓶中加入11mmol 3,5-二羟基苯硼酸和10mmol 4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺,以及20mmol无水碳酸钠,并加入40ml无水甲苯和10ml水,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol四(三苯基膦)钯,随后继续抽真空通
氮气三次,然后在100℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物(4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺基)-1,3-二羟基苯酚。
[0073]1h nmr(500mhz,dmso-d6)δ8.71(d,j=7.5hz,1h),8.67(d,j=7.5hz,1h),7.90(d,j=7.5hz,1h),7.85(d,j=7.5hz,1h),7.63
–
7.57(m,2h),7.30
–
7.23(m,4h),7.21
–
7.16(m,2h),7.13
–
7.04(m,6h),7.02(d,j=1.5hz,2h),6.25(t,j=1.4hz,1h)。
[0074]
(3)聚合物发光材料的合成
[0075][0076]
式中x=0.1,z=0.4,y=0.5。
[0077]
在干燥洁净的三口烧瓶中加入2mmol(4-(8-溴-喹喔啉-5-苯基)-n,n-二苯胺基)-1,3-二羟基苯酚和10mmol 1,6-己二异氰酸酯(hdi),升温至75℃搅拌两小时,随后加入8mmol 2,2-二羟甲基丙酸(dmpa)扩链,升温至85℃反应,随后间隔2小时点板观察hdi浓度,当hdi浓度降到最低时,加入丙酮降低粘度并用三乙胺淬灭hdi,将得到的反应溶液倒入甲醇中搅拌25min析出沉淀,过滤得到黄色固体;将黄色固体加入2mol/l的盐酸溶液中搅拌溶解,然后以氯仿萃取,分离并收集有机相;向有机相中加入去离子水,搅拌30min,萃取分离,重复进行3次,收集剩余有机相;将有机相经真空旋转蒸发浓缩,浓缩液倒入甲醇中搅拌30min析出沉淀,过滤得到黄色固体,然后将固体置于索氏提纯器中,以丙酮提纯40h后,于70℃真空干燥箱中干燥6h,得到以三苯胺和喹喔啉作为给受体组合的聚合物发光材料。
[0078]
本实施例得到的聚合物发光材料的数均分子量为5026,重均分子量为7690,分散度为1.53。
[0079]
图1为实施例1制备的聚合物发光材料在甲苯中的荧光光谱图,由图1可以看出,聚合物发光材料在甲苯溶液中发绿光,发光峰位于528nm。
[0080]
实施例2
[0081]
以三苯胺和二氰基喹喔啉作为给受体组合的聚合物发光材料的合成
[0082]
(1)6-溴-7-(4-二苯胺基)-2,3-二氰基喹喔啉的合成
[0083]
[0084]
在干燥洁净的三口烧瓶中加入11mmol 4-硼酸三苯胺和10mmol 6,7-二溴-2,3-二氰基喹喔啉,以及20mmol无水碳酸钠,并加入40ml无水甲苯和10ml水,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol四(三苯基膦)钯,随后继续抽真空通氮气三次,然后在100℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物6-溴-7-(4-二苯胺基)-2,3-二氰基喹喔啉。
[0085]1h nmr(500mhz,dmso-d6)δ8.63(s,1h),8.44(s,1h),7.61
–
7.55(m,2h),7.30
–
7.22(m,6h),7.13
–
7.07(m,4h),7.06(tt,j=7.4,1.5hz,2h)。
[0086]
(2)6-(3,5-二羟基苯)-7-(4-二苯胺基)-2,3-二氰基喹喔啉的合成
[0087][0088]
在干燥洁净的三口烧瓶中加入11mmol 3,5-二羟基苯硼酸和10mmol 6-溴-7-(4-二苯胺基)-2,3-二氰基喹喔啉,以及20mmol无水碳酸钠,并加入40ml无水甲苯和10ml水,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol四(三苯基膦)钯,随后继续抽真空通氮气三次,然后在100℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物6-(3,5-二羟基苯)-7-(4-二苯胺基)-2,3-二氰基喹喔啉。
[0089]1h nmr(500mhz,dmso-d6)δ8.59(s,1h),8.54(s,1h),7.68
–
7.62(m,2h),7.31
–
7.24(m,4h),7.27
–
7.18(m,2h),7.13
–
7.03(m,6h),6.90(d,j=1.5hz,2h),6.27(t,j=1.4hz,1h)。
[0090]
(3)聚合物发光材料的合成
[0091][0092]
式中x=0.1,z=0.4,y=0.5。
[0093]
在干燥洁净的三口烧瓶中加入2mmol 6-(3,5-二羟基苯)-7-(4-二苯胺基)-2,3-二氰基喹喔啉和10mmol 1,6-己二异氰酸酯(hdi),升温至75℃搅拌两小时,随后加入8mmol 2,2-二羟甲基丙酸(dmpa)扩链,升温至85℃反应,随后间隔2小时点板观察hdi浓度,当hdi浓度降到最低时,加入丙酮降低粘度并用三乙胺淬灭hdi,将得到的反应溶液倒入甲醇中搅拌25min析出沉淀,过滤得到红色固体;将红色固体加入2mol/l的盐酸溶液中搅拌溶解,然后以氯仿萃取,分离并收集有机相;向有机相中加入去离子水,搅拌20min,萃取分离,重复进行3次,收集剩余有机相;将有机相经真空旋转蒸发浓缩,浓缩液倒入甲醇中搅拌30min析
出沉淀,过滤得到红色固体,然后将固体置于索氏提纯器中,以丙酮提纯40h后,于70℃真空干燥箱中干燥6h,得到以三苯胺和二氰基喹喔啉作为给受体组合的聚合物发光材料。
[0094]
本实施例得到的聚合物发光材料的数均分子量为5497,重均分子量为8190,分散度为1.49。
[0095]
图2为实施例2制备的聚合物发光材料在甲苯中的荧光光谱图,由图2可以看出,聚合物发光材料在甲苯溶液中发红光,发光峰位于635nm。
[0096]
实施例3
[0097]
以9,9-二甲基-9,10二氢吖啶和喹喔啉作为给受体组合的聚合物发光材料的合成
[0098]
(1)10-(8-溴-喹喔啉基-5)-9,9-二甲基-9,10-二氢吖啶的合成
[0099][0100]
在干燥洁净的三口烧瓶中加入11mmol 9,9-二甲基-9,10-二氢吖啶和10mmol 1,4-二溴喹喔啉,以及20mmol无水叔丁醇钠,并加入40ml无水甲苯,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol醋酸钯和1mmol(三叔丁基)膦四氟硼酸盐,随后继续抽真空通氮气三次,然后在110℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物10-(8-溴-喹喔啉-5)-9,9-二甲基-9,10-二氢吖啶。
[0101]1h nmr(500mhz,dmso-d6)δ8.64(d,j=7.5hz,1h),8.59(d,j=7.5hz,1h),7.93(d,j=7.5hz,1h),7.45(d,j=7.5hz,1h),7.19(ddd,j=7.9,6.6,2.0hz,2h),7.17
–
7.08(m,6h),1.57(s,4h)。
[0102]
(2)5-(8-(9,9-二甲基-9,10-二氢吖啶)-喹喔啉基-5)-1,3-二羟基苯酚的合成
[0103][0104]
在干燥洁净的三口烧瓶中加入11mmol 3,5-二羟基苯硼酸和10mmol 10-(8-溴-喹喔啉-5)-9,9-二甲基-9,10-二氢吖啶,以及20mmol无水碳酸钠,并加入40ml无水甲苯和10ml水,随后将三口烧瓶抽真空通氮气,在氮气保护下加入0.5mmol四(三苯基膦)钯,随后继续抽真空通氮气三次,然后在100℃下反应12小时,之后过滤,萃取,旋干之后进行柱层析得到产物5-(8-(9,9-二甲基-9,10-二氢吖啶)-喹喔啉基-5)-1,3-二羟基苯酚。
[0105]1h nmr(500mhz,dmso-d6)δ8.62(d,j=7.5hz,1h),8.55(d,j=7.5hz,1h),7.86(d,j=7.5hz,1h),7.49(d,j=7.5hz,1h),7.22
–
7.17(m,2h),7.17
–
7.09(m,4h),7.08(ddd,j=7.1,5.4,3.7hz,2h),6.95(d,j=1.4hz,2h),6.26(t,j=1.5hz,1h),1.57(s,4h)。
[0106]
(3)聚合物发光材料的合成
[0107][0108]
式中x=0.2,z=0.3,y=0.5。
[0109]
在干燥洁净的三口烧瓶中加入4mmol 5-(8-(9,9-二甲基-9,10-二氢吖啶)-喹喔啉基-5)-1,3-二羟基苯酚和10mmol 1,6-己二异氰酸酯(hdi),升温至75℃搅拌两小时,随后加入6mmol 2,2-二羟甲基丙酸(dmpa)扩链,升温至85℃反应,随后间隔2小时点板观察hdi浓度,当hdi浓度降到最低时,加入丙酮降低粘度并用三乙胺淬灭hdi,将得到的反应溶液倒入甲醇中搅拌25min析出沉淀,过滤得到橙色固体;将橙色固体加入2mol/l的盐酸溶液中搅拌溶解,然后以氯仿萃取,分离并收集有机相;向有机相中加入去离子水,搅拌20min,萃取分离,重复进行3次,收集剩余有机相;将有机相经真空旋转蒸发浓缩,浓缩液倒入甲醇中搅拌25min析出沉淀,过滤得到橙色固体,然后将固体置于索氏提纯器中,以丙酮提纯40h后,于70℃真空干燥箱中干燥6h,得到以9,9-二甲基-9,10-二氢吖啶和喹喔啉作为给受体组合的聚合物发光材料。
[0110]
本实施例得到的聚合物发光材料的数均分子量为4699,重均分子量为7330,分散度为1.56。
[0111]
图3为实施例3制备的聚合物发光材料在甲苯中的荧光光谱图,由图3可以看出,聚合物发光材料在甲苯溶液中发橙光,发光峰位于604nm。
[0112]
应用例
[0113]
将实施例1制备的聚合物发光材料以ito/pedot:pss(厚度为40nm)/实施例1制备的聚合物发光材料(厚度为40nm)/tmpypb(厚度为50nm)/lif(厚度为1nm)/al(厚度为150nm)的结构经过湿法制备为oled器件,其发光颜色为橙光,发光光谱范围为500~600nm,光致发光量子产率为85%,最大外量子效率为15%。
[0114]
将实施例2制备的聚合物发光材料以ito/pedot:pss(厚度为40nm)/实施例2制备的聚合物发光材料(厚度为40nm)/tmpypb(厚度为50nm)/lif(厚度为1nm)/al(厚度为150nm)的结构经过湿法制备为oled器件,其发光颜色为红光,发光光谱范围为600~700nm,光致发光量子产率为81%,最大外量子效率为12%。
[0115]
将实施例3制备的聚合物发光材料以ito/pedot:pss(厚度为40nm)/实施例3制备的聚合物发光材料(厚度为40nm)/tmpypb(厚度为50nm)/lif(厚度为1nm)/al(厚度为150nm)的结构经过湿法制备为oled器件,其发光颜色为红光,发光光谱范围为550~700nm,光致发光量子产率为79%,最大外量子效率为13%。
[0116]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。