1.本发明属于分子生物学技术领域,具体涉及一种急性髓系白血病检测探针组合物及其应用。
背景技术:
2.急性髓系白血病(acute myeloid leukemia,aml)是一组异质性的侵袭性血液系统恶性肿瘤,其临床病程和治疗反应存在广泛差异。急性髓系白血病的特征是未成熟的骨髓前体的扩张,从而导致正常的造血功能受损,最终导致骨髓衰竭。它是由影响造血干细胞或祖细胞增殖和/或分化的遗传改变的逐步积累引起的。aml在发生时常伴随着高度异质的细胞遗传学和分子生物学异常,这些异常能够辅助诊断、评估预后,从而参与治疗决策。近年来,测序技术的进步使得急性髓系白血病(aml)复发突变的鉴定成为可能。通过aml的测序工作,一个广泛的体细胞突变目录逐步呈现出来。其中反复发生的突变显示出高度的重叠,而频率较低的突变则高度分化。这表明,反复发生的突变是导致疾病的真正致癌驱动突变,而低频突变则可能是不影响aml生物学的无关突变。
3.人类aml患者中发现的最常见的异常之一是在正常造血发育过程中起作用的转录因子和表观遗传调节因子的突变,包括融合蛋白(如:pml-rara、runx1-runx1t1、cbfb-myh11等)和aml相关基因的重要突变(如:tp53、cebpa、flt3、dnmt3a等)等。如,专利cn107964561a公开了检测cebpa基因突变的引物及检测方法,进一步用于判断正常核型aml患者的预后情况,用于检测的准确度、特异性和灵敏度较高。专利cn108929912a则公开了一种用于筛查急性髓系白血病的引物、试剂盒以及方法,其包括用于扩增人ccn1基因的snp位点rs2297141和rs6576776片段的引物组合物,通过对位点rs2297141,rs6576776单个核酸多态性检测与分析,实现汉族人群急性髓系白血病的早期筛查和易感性的风险评估。然而,现有的这些评估标志物并不能完全有效地预测aml患者的风险,原因如下:第一,约50%的aml患者核型正常,既无基因组结构变异也无已知的基因突变,这些患者的复发风险和生存时间用现有的标志物难以预测。第二,虽然有些基因突变对生存时间的分类效果在统计学上极其显著,但是这些基因的突变率极低,绝大部分患者中无该类基因的突变,因此无法根据基因突变预测这些患者的生存时间。第三,现有的评估标准(如:european leukemia net guideline)通常将患者归为高、中、低风险三类,绝大部分患者被归为中风险类,进而采用相似的治疗手段。然而,被归类为中风险类的患者对于联合化疗的治疗反应有好有坏,这表明,对于中风险类的患者,还应使用新的预后标志物进行细分,进而调整治疗方案,以达到更好的治疗效果。第四,不同的基因突变间往往存在共现或互斥的情况,因此很多现有的预测因子没有独立的预后效果。比如,npm1突变且flt3不突变的患者具有较好的预后。
4.因此,亟需提供一种急性髓系白血病检测灵敏度、准确度更高的探针组合物及其检测方法。
技术实现要素:
5.为了解决现有技术中的上述问题,本技术提供了一种急性髓系白血病aml相关基因群检测捕获探针组合物及其应用。采用本发明所述的捕获探针组合物检测aml相关基因群,具有较好的灵敏度和准确度,扩增效率高,且操作简便,快速省时。
6.为了实现上述发明目的,本发明提供如下技术方案:
7.一方面,本发明提供了一种急性髓系白血病相关基因群检测的探针组合物,所述的探针组合物包括seq id no:1-seq id no:538所示的序列。
8.具体地,所述的探针组合物用于检测下表1所示的基因及热点突变位点:
9.表1.基因及热点突变位点
10.11.12.13.14.15.16.[0017][0018]
注:nonsense,无义突变,突变可造成氨基酸编码提前终止,进而可能影响蛋白功能;frameshift,移码突变,突变使翻译阅读框发生改变,从而导致氨基酸序列发生变化;splice site,剪接位点突变,经数据库预测可能影响剪接位点,造成转录产物异常。
[0019]
另一方面,本发明提供了上述探针组合物在制备急性髓系白血病相关基因群检测产品中的应用。
[0020]
具体地,所述的产品包括但不限于独立试剂、芯片或试剂盒。
[0021]
又一方面,本发明提供了一种急性髓系白血病相关基因群检测的产品,所述的产品包括上述探针组合物。
[0022]
具体地,所述的产品包括但不限于独立试剂、芯片或试剂盒。
[0023]
又一方面,本发明提供了上述探针组合物或产品在检测急性髓系白血病相关基因群突变中的应用。
[0024]
又一方面,本发明提供了一种检测急性髓系白血病相关基因群突变的方法,所述的方法为非疾病诊断和治疗方法,所述的方法包括利用上述探针组合物或产品对待测样本中急性髓系白血病相关基因群的突变进行检测。
[0025]
具体地,所述的方法包括以下步骤:
[0026]
(1)血液样本单个核细胞分离;
[0027]
(2)样本dna提取;
[0028]
(3)文库构建;
[0029]
(4)上机测序;
[0030]
(5)数据分析。
[0031]
与现有技术相比,本发明提供的试剂盒具有如下有益效果:
[0032]
(1)采用本发明提供的探针组合物检测aml(急性髓系白血病)相关基因群,具有较好的灵敏度和准确度,扩增效率高,且操作简便,快速省时。
[0033]
(2)利用本发明提供的探针组合物,可以实现在骨髓形态学和血常规检查之外对aml进行辅助诊断,特别是对于疾病后续的治疗方向和效果做出预判具有特殊的优势,可弥补临床aml在分子诊断、疾病演变、治疗、预后、用药等指导方面的不足。
附图说明
[0034]
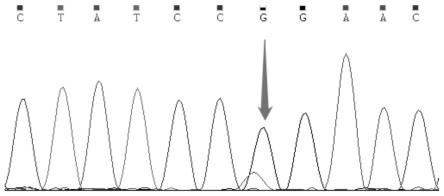
图1为idh2:p.r140q sanger测序峰图。
[0035]
图2为dnmt3a:p.r882p sanger测序峰图。
[0036]
图3为npm1:p.w288cfs*12sanger测序峰图。
[0037]
图4为gata1:p.h71r sanger测序峰图。
[0038]
图5为tet2:p.h1778r sanger测序峰图。
具体实施方式
[0039]
下面结合具体实施例,对本发明作进一步详细的阐述,下述实施例不用于限制本发明,仅用于说明本发明。以下实施例中所使用的实验方法如无特殊说明,实施例中未注明具体条件的实验方法,通常按照常规条件,下述实施例中所使用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0040]
实施例中未注明具体技术或条件者,均按照本领域内的文献所描述的技术或条件(如参考j.萨姆布鲁克等著,黄培堂等译的《分子克隆实验指南》,第三版,科学出版社)或者按照产品说明书进行。
[0041]
1.试剂及仪器
[0042]
(1)本技术所用的试剂信息如下表2所示:
[0043]
表2.试剂
[0044][0045]
(2)本技术所用的仪器信息如下表3所示:
[0046]
表3.仪器
[0047][0048]
实施例1.一种aml相关基因群检测的探针组合物及其试剂盒
[0049]
(1)所述的探针组合物包括seq id no:1-seq id no:538所示的序列。
[0050]
所述的探针组合物用于检测上表1所示的基因的热点突变(共786个)。
[0051]
实施例2.一种aml相关基因群检测的方法
[0052]
一、血液标本单个核细胞分离
[0053]
准备15ml无菌塑料离心管放在试管架上并打开盖子,加入6-7ml淋巴细胞分离液;把抗凝标本(若标本量少于1ml,加入2ml盐水稀释颠倒混匀)缓慢倒入装有淋巴细胞分离液的15ml无菌塑料离心管中并盖上盖子,同时标记标本编号;然后离心(4℃2000r/min 15min);准备15ml无菌塑料离心管放在相应试管架上并打开盖子,加入5ml无菌生理盐水;然后用一次性无菌塑料吸管充分吸取离心后的标本白细胞环并加入到盛有5ml无菌生理盐水的离心管中,同时标记标本编号;所有标本配平、离心(4℃1500r/min 5min);小心弃上清,向15ml无菌塑料离心管中加入9-10ml无菌生理盐水;离心(4℃1500r/min5min);小心弃上清,用新吸管把管底标本白细胞沉淀吸入到1.5ml无菌塑料离心管中,并用gs缓冲液补足体积到0.25ml,同时标记标本编号,准备用于dna的提取。
[0054]
二、血液标本dna提取
[0055]
向含有标本白细胞层的1.5ml经高压灭菌塑料离心管中加入20μl蛋白酶k(蛋白酶k可以提前加入到1.5ml无菌塑料离心管中);加入200μl gb缓冲液,盖好盖子,充分混匀15秒;放入水浴锅中,56℃孵育20分钟;取出离心管,小心开盖,加入200μl无水乙醇,盖好盖子,充分混匀15秒;转移到套有收集管的吸附柱cg2中,吸附柱cg2柱上标记标本编号,12000转/分钟离心2分钟;取出吸附柱cg2,换一新收集管,打开吸附柱cg2的盖子,向吸附柱cg2中加入500μl gdb缓冲液,盖好盖子,12000转/分钟离心1分钟;取出吸附柱cg2,再换一新收集管,打开吸附柱cg2的盖子,向吸附柱cg2中加入600μl pwb缓冲液,12000转/分钟离心1分钟;重复步骤上述步骤一次。取出吸附柱cg2,再换一新收集管,12000转/分钟离心2分钟;将吸附柱cg2置于一新1.5ml离心管中,在实验台开盖静置5分钟;加入65μl eb缓冲液,室温下孵育5分钟,12000转/分钟离心2分钟;1.5ml离心管内容物即为标本的dna,在1.5ml离心管上标记好标本编号并盖好盖子(贴标签;将提取得到的dna涡旋离心,测nanodrop浓度和qubit浓度,将浓度更新到标本信息表里,给dna编号,暂放于4℃已待后续实验使用。
[0056]
三、文库构建:
[0057]
1、样本前处理:
[0058]
组织dna样本,若样本中无edta取所需量的组织dna,用nuclease-free water(nf water)稀释到35μl。如果样本中有高浓度edta建议2倍体积的xp磁珠纯化后再建库。
[0059]
2、样本酶切打断:
[0060]
全程冰上加入试剂kapa frag buffer(10
×
)-5μl、kapa frag enzyme-10μl、稀释后样本-35μl。体系共50μl,涡旋混匀。具体反应程序如下表4:
[0061]
表4
[0062][0063]
反应结束后将离心管取出放置于冰上,每个反应加入5μl stop solution;向每个反应中加入110μl已室温平衡的xp beads(2
×
),涡旋混匀,室温孵育5min;瞬时离心,将ep管置于磁力架上吸附5min,弃上清;加入200μl新配的80%乙醇,静置30s,弃上清;重复上步一次,80%乙醇共洗2次;瞬时离心,用10μl枪头吸净液体,室温干燥3-5min;加入42μl的nfwater,重悬磁珠,室温孵育5min;瞬时离心,置于磁力架上直至液体澄清,吸取40μl上清至一新的pcr管中。
[0064]
3、末端修复&加a:
[0065]
反应体系:end repair&a-tailing buffer-6μl、end repair&a-tailing enzyme-4μl、上步反应产物-40μl。体系50μl,涡旋混匀。
[0066]
反应条件:20℃-30min,65℃-30min,4℃-hold(特别注意:热盖70℃)。
[0067]
4、接头连接:
[0068]
反应体系:end repair and a-tailing reaction product-50μl、idt udi接头(15μm)-2μl,涡旋混匀,ligation buffer-26μl、dna ligase-2μl。体系80μl,涡旋混匀。
[0069]
反应条件:放置于pcr仪上,20℃孵育20min(lid open)(有条件可适当延迟反应时
间)。
[0070]
将反应管瞬时离心,吸取全部反应液于含有64μl已室温平衡的sp beads(0.8
×
)的1.5ml ep管中,涡旋混匀,室温孵育5-10min;瞬时离心,将ep管置于磁力架上吸附5min,弃上清;加入200μl新配的80%乙醇,静置30s,弃上清;重复上步一次,80%乙醇共洗2次;瞬时离心,用10μl枪头吸净液体,室温干燥3-5min;加入22μl的nfwater,重悬磁珠,室温孵育5min;瞬时离心,置于磁力架上直至液体澄清,吸取20μl上清至一新的pcr管中。
[0071]
5、文库富集:
[0072]
反应程序如下表5所示。
[0073]
表5
[0074][0075]
将反应管瞬时离心,吸取全部反应液于含有40μl已室温平衡的sp beads(0.8
×
)的1.5ml ep管中,涡旋混匀,室温孵育5min;瞬时离心,将ep管置于磁力架上吸附5min,弃上清;加入200μl新配的80%乙醇,静置30s,弃上清;重复上步一次,80%乙醇共洗2次;瞬时离心,用10μl枪头吸净液体,室温干燥3-5min;加入50μl的nfwater,重悬磁珠,室温孵育5min;瞬时离心,置于磁力架上直至液体澄清,吸取48μl上清至一新的ep管中,做好标记,即为得到的文库;取1μl的文库进行qubit定量,取1μl的文库进行文库片段分析,记录文库浓度与产量。
[0076]
四、杂交捕获:
[0077]
1、dna文库混合:
[0078]
取一个新的1.5ml低吸附离心管,将不同index的文库混合,一个pool最多8个样本一起杂交;每个预文库样本投入300ng。
[0079]
2、封闭文库并干燥,体系如下表6所示:
[0080]
表6
[0081]
封闭反应体系单个用量(μl)dna library300ng/例human cot dna5nadprepnanoblockers2
[0082]
充分混匀,使用提前预热的真空浓缩仪,55℃烘干。
[0083]
3、探针与文库杂交,体系如下表7所示:
[0084]
表7
[0085]
杂交混合液用量(μl)
xgen 2
×
hybridization buffer8.5xgen hybridization buffer enhancer2.7probe探针4nuclease-free water1.8
[0086]
上述试剂加入到烘干的文库中,用移液器吹打混匀,室温孵育5-10min;充分涡旋混匀,瞬时离心;转移上述17μl杂交混合液至新的0.2ml低吸附pcr管中,瞬时离心。
[0087]
反应条件:95℃-30s;65℃-4h;65℃-hold(特别注意:pcr仪热盖100℃)。
[0088]
4、准备缓冲液,具体如下表8所示:
[0089]
表8
[0090]
试剂(试剂和nf water提前平衡30min)母液用量(μl)water(μl)xgen 2
×
bead wash buffer160160xgen 10
×
wash buffer 128252xgen 10
×
wash buffer 216144xgen 10
×
wash buffer 316144xgen 10
×
stringent wash buffer32288
[0091]
每个反应分装110μl xgen 1
×
wash buffer 1至新的pcr管中,65℃孵育,使用前至少孵育15min;每个反应分装310μl xgen 1
×
stringent wash buffer到两个pcr管中,每管155μl,置于65℃孵育,使用前至少孵育15min(置于热盖70℃、65℃孵育的另一pcr仪器中);剩余1
×
缓冲液放置于室温备用。
[0092]
5、准备streptavidin beads:
[0093]
取出streptavidin beads并室温平衡至少30min,涡旋15s充分混匀磁珠;取适量磁珠至新的1.5ml低吸附离心管中,放置在磁力架上带澄清后弃去上清;每个捕获加入100μl xgen 1
×
bead wash buffer,移液器缓慢吹打10次;瞬时离心,放入磁力架,待液体澄清,弃去上清;重复一次,从磁力架取下离心管,每个捕获加入100μl xgen 1
×
bead wash buffer,移液器缓慢吹打10次;瞬时离心,放入磁力架,待液体澄清,弃去上清;再重复一次,从磁力架取下离心管,每个捕获加入100μl xgen 1
×
bead wash buffer,移液器缓慢吹打10次;瞬时离心,放入磁力架待液体澄清,弃去上清;每份磁珠中加入17μl磁珠重悬混合液,充分涡旋混匀,瞬时离心;将17μl磁珠重悬混合液,放置在pcr仪上孵育5min(可选)。
[0094]
磁珠重悬混合液体系如下表9所示。
[0095]
表9
[0096]
磁珠重悬混合液用量(μl)xgen 2
×
hybridization buffer8.5xgen hybridization buffer enhancer2.7nuclease-free water5.8
[0097]
6、磁珠结合dna:
[0098]
杂交反应结束后,从pcr仪上取出含有杂交样本的pcr管;分装17μl重悬磁珠至上述含有杂交样本的pcr管中,充分涡旋混匀,瞬时离心;置于pcr仪上,65℃45min(热盖70℃,待pcr仪热盖温度完全降温至70℃时,再将样本置于pcr仪上)。(每间隔10min,涡旋混匀1次,保持样本处于悬浮状态)。
[0099]
7、清洗杂交捕获文库:
[0100]
从pcr仪取出pcr管,加100μl 65℃孵育的xgen 1
×
washbuffer 1,移液器吹打10次混匀,避免出现气泡;瞬时离心,置于磁力架,待液体澄清,弃去上清;再次瞬时离心,用10μl枪头吸净残液;从磁力架取下离心管,加入150μl65℃孵育的xgen 1
×
stringentwash buffer,移液器吹打10次混匀,注意避免出现气泡;65℃孵育5分钟;盖上热盖;瞬时离心,放入磁力架,待液体澄清,弃去上清;再次瞬时离心,用10μl枪头吸净残液;从磁力架取下离心管,加入150μl65℃孵育的xgen 1
×
stringentwash buffer,移液器吹打10次混匀,注意避免出现气泡;65℃孵育5分钟;盖上热盖;瞬时离心,放入磁力架,待液体澄清,弃去上清;再次瞬时离心,用10μl枪头吸净残液;从磁力架取下离心管,加入150μl xgen 1
×
wash buffer 1,充分涡旋混匀,室温孵育2min,隔30s震荡一次,以保持悬浮状态;瞬时离心,置于磁力架,待液体澄清,弃去上清;从磁力架取下离心管,加入150μl xgen 1
×
wash buffer 2,充分涡旋混匀,室温孵育2min,隔30s震荡一次,以保持悬浮状态;瞬时离心,置于磁力架,待液体澄清,弃去上清;从磁力架取下离心管,加入150μl xgen 1
×
wash buffer 3,充分涡旋混匀,室温孵育2min,隔30s震荡一次,以保持悬浮状态;瞬时离心,置于磁力架,待液体澄清,弃去上清;瞬时离心,用10μl枪头吸净残留的xgen 1
×
wash buffer3,从磁力架取下离心管;加入20μl nuclease-free water,移液器吹打10次混匀,以确保磁珠充分悬浮,转移至含有post-capture pcr mix的0.2ml pcr管,涡旋混匀,轻微瞬时离心,避免磁珠沉淀。
[0101]
8、post-capture pcr:
[0102]
体系如下表10所示。
[0103]
表10
[0104]
post-capture pcr混合液单个用量(μl)2
×
kapa hifihotstartreadymix25kapa primer(5μm/each primer)5杂交捕获文库20
[0105]
反应条件如下表11所示:
[0106]
表11
[0107][0108]
取出ampure xp beads室温平衡至少30min备用;将反应管瞬时离心,吸取全部反应液转入含有75μl(1.5
×
)已室温平衡的纯化beads的1.5ml低吸附离心管中,涡旋混匀,室温孵育5-10min;瞬时离心,离心管置于磁力架上吸附2-5min,至液体澄清,弃去上清;保持离心管位于磁力架上,加入150μl 80%乙醇,静置1min,弃去上清;重复一次,加入150μl 80%乙醇,静置1min,弃去上清;室温干燥1-3min,注意勿让磁珠过度干透;加入50μl eb洗脱,充分混匀磁珠,室温孵育5min;瞬时离心,置于磁力架上,至液体澄清,转移48μl上清至
新的离心管中,做好标记,即为得到的文库;取1μl的文库进行qubit定量,记录文库浓度与产量。
[0109]
五、上机测序
[0110]
1、试剂准备:
[0111]
将试剂夹盒从冰箱-20℃中取出,在冰箱4℃解冻至少18个小时,也可以放入装有室温清水的水槽中解冻60分钟,注意不要将整个夹盒浸泡在水中;解冻后将夹盒翻转五次以混匀试剂,之后抬起夹盒边缘轻敲(左右两边都要需要)以减少气泡。(注:试剂夹盒中的试剂对光敏感,在解冻时注意避光。)将ht1从冰箱-20℃中取出,在室温下解冻。使用时放在冰盒上。将流动槽从冰箱4℃中取出,置于室温下30分钟。缓冲液夹盒在室温储存,在上机时取出即可。试剂夹盒、缓冲液盒、ht1、流动槽的标签揭下,贴在上机信息表上。
[0112]
配制0.2n naoh溶液(0.2mol/l naoh溶液):5μl 10mol/l naoh 45μl h2o涡离后得到50μl 1mol/l naoh溶液。再取20μl 1mol/l naoh溶液 80μl h2o涡离后得到100μl 0.2n naoh溶液。配制0.2mol/l tris-hcl(ph7.0)溶液:20μl 1mol/l tris-hcl 80μl h2o涡离后得到100μl 0.2mol/l tris-hcl(ph7.0)。混pool并稀释至4nmol/l:根据“nextseq上机信息表”先向1.5ml低吸附管中加入ht1,再加入文库。涡离后得到浓度为4nmol/l的pooling文库。
[0113]
2、测定pooling文库浓度:
[0114]
取3个500μl的qubit分析管,每管加入195μl的equalbit 1x dsdna hs working solution待用。再取2个分析管加入190μl的equalbit 1x dsdna hs working solution,分别加入10μl 1号标准品和2号标准品。涡旋离心后定标,若2号标准品的浓度为100ng/μl或99.8ng/μl即为定标通过;若低于99.8ng/μl,定标失败,应重新涡旋离心标准品或重新配制qubit标准品。定标通过后向待用的三管分别加入5μl本次测序的pooling文库、上次测序的pooling文库以及再上次测序的pooling文库。涡旋离心后测定qubit浓度。该浓度将用于计算上机浓度。
[0115]
变性文库:取5μl上述文库,加入到新的1.5ml低吸附管,再向其中加入5μl0.2n naoh溶液,涡离后在室温下变性5min,5min后向其中加入5μl 0.2mol/l tris-hcl(ph7.0),涡离后立即放冰上进行下一步。
[0116]
3、稀释变性文库:
[0117]
先向管中加入985μl ht1,再加入变性文库15μl,涡离后得到1ml 20pmol/l的稀释变性文库。稀释变性文库应放在冰上。
[0118]
计算上机浓度与稀释变性文库加样体积:
[0119][0120][0121]
注:上次测序与再上次测序的数据均可用来计算上机浓度,但计算时只应使用一次测序的数据。
[0122]
4、制备上机文库:
[0123]
根据计算好的稀释变性文库加样体积,先计算所需ht1体积:1300-稀释变性文库
体积;配制上机文库时先加入ht1,再加入稀释变性文库,涡旋离心后即得到总体积为1300μl的上机文库,使用前应放冰上待用。
[0124]
5、加样:
[0125]
用1000μl的枪头刺破试剂夹盒10号孔,向其中加入上机文库,加样时沿管壁加样,注意不要打进气泡,枪头中可以适当残留一些上机文库。
[0126]
6、上机测序:
[0127]
更换流动槽:打开流动槽的锡纸包装,打开塑料盒包装,如果流动槽表面有灰尘或污渍,用无尘纸蘸取无水乙醇轻轻擦拭流动槽表面。取下清洗用的流动槽,安装上新的流动槽。
[0128]
更换缓冲液盒以及清理废液盒:取下清洗用缓冲液盒,清空液体,倒扣在吸水纸上。换上新的缓冲液盒。清空废液盒中的废液,重新放回舱内。
[0129]
更换试剂夹盒:从左侧舱取出清洗夹盒,清空液体,倒扣在吸水纸上。把已上样的试剂夹盒放入舱内。
[0130]
复查运行设定与仪器自检:再次确认运行设定后选择“next”,等待仪器完成自检,自检完成后选择“start”开始测序。
[0131]
六、数据分析:
[0132]
1、原始测序信号文件利用bcl2fastq程序转化成fastq格式文件;原始数据(fastq格式文件)经过fastp软件质控,过滤掉低质量、不确定碱基、含有接头的reads得到clean data(fastq格式文件);利用bwa软件将质控合格的clean data比对到人类hg19参考基因组,得到bam格式文件;利用sambamba软件去除bam文件中的duplicate reads;利用vardcct软件对bam格式文件进行变异检测,得到vcf格式文件;利用annovar软件进行相关数据库注释(注释内容包括突变在基因组中的位置,关联的基因,基因外显子编号,核苷酸水平变异,对应的蛋白水平变异,该突变在dbsnp(http://www.ncbi.nlm.nih.gov/snp/),1000genomes数据库(http://www.1000genomes.org/)和癌症突变数据库cosmic(http://cancer.sanger.ac.uk/cosmic)中的注释,polyphen(http://genetics.bwh.harvard.edu/pph2/)和sift(http://sift.jcvi.org/)功能预测结果等)。
[0133]
2、按照以下原则筛选突变位点:
[0134]
(1)根据查阅文献指南等整理出的热点库进行注释,按照不同分级对热点突变判读为致病突变;
[0135]
(2)根据突变所在的基因组位置和类型,过滤掉对蛋白产物序列不产生影响的突变;
[0136]
(3)利用1000genomes数据,注释每个突变在人群中的比例,如果比例小于等于1%则不认为是多态性位点;
[0137]
(4)经过(3)后,一个突变满足“非多态性”且为特殊突变(无义/移码突变/插入/缺失(包括非移码突变)/经典剪接位点(
±
1/2时)/起始密码丢失/终止密码子丢失),则判读为与疾病可能相关;
[0138]
(5)检索癌症数据库cosmic,查询一个突变在cosmic中是否有记载,其出现在血液系统肿瘤中;
[0139]
(6)利用蛋白质功能预测软件polyphen-2等数据库预测该突变是否影响蛋白功
能;
[0140]
(7)如果经过(3)(5)(6)后,一个突变满足“非多态性”、“在血液系统肿瘤中记载过”和“影响蛋白功能”这三条中至少两条的,将被判为意义不明突变。
[0141]
实验例1.重复性检测
[0142]
根据实施例1提供的探针组合物,分别采用实施例2的检测方法检测aml患者样本2例,重复检测三次,其结果如下表12所示。
[0143]
表12.重复性检测结果
[0144][0145]
由上表可知,本技术所述的探针组合物具有较好的重复性。
[0146]
实验例2.灵敏度检测
[0147]
根据实施例1提供的探针组合物,分别采用实施例2的检测方法提取的不同浓度的dna样本进行检测。选择已知突变频率的阳性突变位点,按一定浓度梯度等比稀释样本(1;1:2;1:4;1:8;1:16;1:32;1:64),并保证总核酸量保持不变,常规实施该项目检测,其结果如下表13所示。
[0148]
表13.灵敏度检测结果
[0149][0150]
由上表可知,本技术所述的探针组合物具有较好的灵敏性。
[0151]
实验例3.准确性检测
[0152]
选取临床样本2例,采用本技术所述的检测方法进行检测,与sanger测序结果(临床结果)进行比较,检测结果如下表14,sanger测序峰图如图1-5所示。
[0153]
表14.准确性检测结果
[0154]
突变位点变异频率sanger测序idh2:p.r140q9.6%,阳性阳性dnmt3a:p.r882p43.7%,阳性阳性npm1:p.w288cfs*1244.0%,阳性阳性gata1:p.h71r46.1%,阳性阳性tet2:p.h1778r62.0%,阳性阳性
[0155]
由上表可知,本技术所述的探针组合物具有较好的准确性。
[0156]
本实施例sanger测序所用的引物序列如下表15所示:
[0157]
表15
[0158][0159]
上述实验均经过中心伦理委员会批准,并经患者同意。
[0160]
以上仅为本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些均属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。