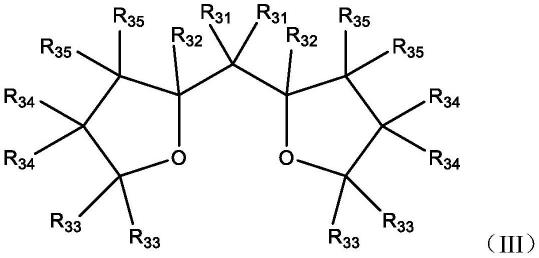
1.本发明属于有机光电材料领域,具体涉及荧蒽类化合物及其在有机光电器件中的应用。
背景技术:
2.有机电致发光二极管(oled)因其具有自发光、固态、可弯折、高效率、高像素密度等特点广泛在各类显示装置中使用,例如手机手表等消费品用显示、医疗显示、车载显示、工业控制显示和vr/ar等。oled器件的生产方法主要有溶液法和固体真空蒸发法,目前常用的是固体真空蒸发法,即在一个具有高真空的腔体内加热将材料蒸发到器件用的基板上,但这个过程一般需要数百小时,很多材料的热稳定性差,使用过程纯度变差,从而降低oled器件的使用性能。
3.荧蒽类化合物可用于oled器件,但由于荧蒽具有很大的共轭平面,分子之间的作用力大,导致这类化合物在固体真空蒸发过程中存在真空蒸镀温度高、材料易分解等问题,很大程度地限制其应用。已有报道采用烷基或者杂环烷基取代的方式对荧蒽类化合物的分子结构进行改进,但烷基或者杂环烷基取代氢后,分子结构中芳基-烷基的键振动较大,烷基可自由旋转,导致化合物的稳定性差,从而影响oled器件的整体使用性能。
技术实现要素:
4.本发明提供一种荧蒽类化合物,其化学结构如式(1)所示:
[0005][0006]
式(1)中,*表示荧蒽基团的碳原子,至少两个相邻的*号碳原子与式(2)或式(3)所示的*号碳原子重合形成非芳香环;
[0007]
式(2)和(3)中,b1–
b7各自独立选自o、s、-cr3r
4-或-nr
5-,b1–
b4或b5–
b7之间任意两个基团可连接成环,r3–
r5分别独立选自氢、氘、取代或未取代的c1
–
c6的烷基或含杂原子的烷基、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基;
[0008]
a选自取代或未取代的c6
–
c60芳基、取代或未取代的c5
–
c60杂芳基;
[0009]
或者a选自-ran(rbrc),ra与*连接,选自取代或未取代的c6
–
c30亚芳基、取代或未取代的c5
–
c30亚杂芳基,rb和rc分别独立选自氢、氘、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基、或者ra、rb和rc任意两个连接成环;
[0010]
或者a选自-n(rdre),n与*相连,rd和re分别独立选自氢、氘、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基、或者rd和re连接成环、或者rd或re的碳原子与*相连;
[0011]
a的杂原子选自o、n、s、p、si、se或b;
[0012]
r1分别独立选自氢、氘、氟、三氟化碳基、氰基、硝基、取代或未取代的c1
–
c10直链或支链烷基、取代或未取代的c1
–
c10杂烷基、取代或未取代的c3
–
c20环烷基、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、或者两个或多个相邻的基团彼此连接成环;
[0013]
m选自0
–
7的整数。
[0014]
本发明还提供有机光电器件,其包括第一电极、第二电极、有机层和光学耦合层,其中所述有机层和/或光学耦合层包含所述荧蒽类化合物中的一种或多种。
[0015]
本发明还提供显示或照明装置,其包括所述有机光电器件。
[0016]
与现有技术相比,本发明的有益效果在于:本发明的荧蒽类化合物在荧蒽基团上连接环烷基或杂环烷基,通过环烷基或杂环烷基限制分子结构中芳基-烷基的转动或振动,可以很好地提升化合物的热稳定性,用作有机光电器件的有机层和/或光学耦合层可以降低加工过程中的真空蒸镀温度,从而提高器件的综合性能,延长使用寿命。
附图说明
[0017]
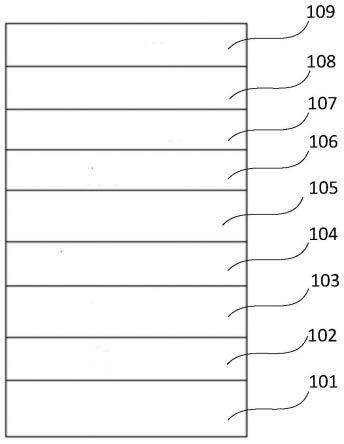
图1是实施例中底部发光有机光电器件的结构示意图。
[0018]
图2是实施例中顶部发光有机光电器件的结构示意图。
[0019]
附图标记如下:101基底层;102-第一电极(阳极);103空穴注入层;104第一层空穴传输层;105第二层空穴传输层;106有机发光层;107空穴阻挡层;108电子传输层;109第二电极(阴极);110覆盖层。
具体实施方式
[0020]
以下详细说明具体公开的化合物及其在有机光电器件中的应用的实施方式。本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0021]
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
[0022]
以下实施例中采用的荧蒽类化合物,其化学结构如式(1)所示:
[0023][0024]
式(1)中,*表示荧蒽基团的碳原子,至少两个相邻的*号碳原子与式(2)或式(3)所示的*号碳原子重合形成非芳香环;
[0025]
式(2)和(3)中,b1–
b7各自独立选自o、s、-cr3r
4-或-nr
5-,b1–
b4或b5–
b7之间任意两个基团可连接成环,r3–
r5分别独立选自氢、氘、取代或未取代的c1
–
c6的烷基或含杂原子的烷基、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基;
[0026]
a选自取代或未取代的c6
–
c60芳基、取代或未取代的c5
–
c60杂芳基;
[0027]
或者a选自-ran(rbrc),ra与*连接,选自取代或未取代的c6
–
c30亚芳基、取代或未取代的c5
–
c30亚杂芳基,rb和rc分别独立选自氢、氘、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基、或者ra、rb和rc任意两个连接成环;
[0028]
或者a选自-n(rdre),n与*相连,rd和re分别独立选自氢、氘、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、取代或未取代的c6
–
c30胺基、或者rd和re连接成环、或者rd或re的碳原子与*相连;
[0029]
a的杂原子选自o、n、s、p、si、se或b;
[0030]
r1分别独立选自氢、氘、氟、三氟化碳基、氰基、硝基、取代或未取代的c1
–
c10直链或支链烷基、取代或未取代的c1
–
c10杂烷基、取代或未取代的c3
–
c20环烷基、取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基、或者两个或多个相邻的基团彼此连接成环,m选自0
–
7的整数。
[0031]
一些实施方式中,式(1)所示的化合物选自以下化学结构中的一种:
[0032][0033]
式(1
–
1)、(1
–
2)和(1
–
3)中,*表示同上;
[0034]
l1选自单键或者以下基团中的一种或两种以上组合:
[0035]
y1–
y5、r1、r2各自独立选自氢、氘、氟、三氟化碳基、氰基、硝基或者以下基团中的一种或两种以上组合:
[0036][0037][0038]
ar选自取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基;
[0039]
a1和a2分别独立选自氢、氘、氟、-cf3、-cn、-no2、取代或未取代的c6
–
c20芳基、取代或未取代的c5
–
c20杂芳基;
[0040]
x选自o、s或-nr
f-,rf选自取代或未取代的c6
–
c30芳基、取代或未取代的c5
–
c30杂芳基;
[0041]
x1–
x7各自独立选自o、s或-crfrg,rf和rg各自独立选自氢、氘、氟、-cf3、-cn、-no2、取代或未取代的c1
–
c10直链或支链烷基、或者取代或未取代的c3
–
c20环烷基;
[0042]
z1–
z8各自独立选自c或n,m选自0
–
7的整数。
[0043]
一些实施方式中,式(1)中非芳香环选自取代或未取代的c5
–
c12环烷基、取代或未取代的c3
–
c10杂环烷基或者以下基团中的一种:
[0044][0045]
上述任意相邻两个*号碳原子位置与式(1)中任意相邻两个*号碳原子位置重合。
[0046]
一些实施方式中,式(1)所示的荧蒽类化合物选自以下化学结构中的一种或多种:
[0047]
[0048]
[0049]
[0050][0051][0052]
合成实施例
[0053]
(a)通过以下反应通式1制备荧蒽类中间体,反应通式1的技术路线如下:
[0054][0055]
(b)通过反应通式2制备化合物反应通式2的技术路线如下:
[0056][0057]
(c)通过反应通式3制备化合物反应通式3的技术路线如下:
[0058][0059]
(d)通过反应通式4和5制备化合物反应通式4和5的技术路线如下:
[0060]
(d-1)反应通式4:
[0061][0062]
(d-2)反应通式5:
[0063][0064]
合成实施例1
[0065][0066]
m1-3的合成:在三口烧瓶,依次加入m1-1(30g,145mmol)、m1-2(37.8g,145mmol)、k2co3(60g,436mmol)、醋酸钯pd(oac)2(0.33g,1.45mmol)、三苯基膦pph3(3.7g,14.5mmol)再加入200ml甲苯中,反应液升温至110℃,搅拌反应6小时。反应液冷却后,加入100ml盐水萃取,有机相用无水硫酸钠干燥,然后真空浓缩,得到的固体用甲苯和石油醚作为冲洗液进行柱色谱分离2次。然后再进行甲苯重结晶2次得到固体,固体烘干,获得m1-3中间体化合物29g,收率68%,lcms(esi离子源)测得m/z:295.0。
[0067]
m1-4的合成:在三口烧瓶,将m1-3(20g,67.8mmol)溶解在200ml的乙腈中,室温下加入三乙胺et3n(27g,271.2mmol),然后将溶液冷却至0℃,滴加全氟丁基磺酰氟nff,然后升温至室温搅拌20小时,用1mol/l的盐酸中和至中性,用乙酸乙酯进行萃取两次,无水硫酸钠干燥后,用甲苯和石油醚作为冲洗液进行柱色谱分离,干燥后得到固体m1-4中间体化合物28g,收率72%。
[0068]
m1的合成:在三口烧瓶,依次加入200ml dma溶剂,m1-3(20g,34.6mmol)、三(二亚苄基丙酮)二钯(pd2(dba)3(1.6g,1.7mmol),2-双环己基膦-2',6'-二甲氧基-1,1'-二联苯sphos(2.8g,6.9mmol),k3po4(29.4g,138mmol),加热110℃,搅拌反应20小时。反应液冷却后,用1mol/l的盐酸中和至中性,用乙酸乙酯进行萃取三次,合并有机相,用无水硫酸钠干燥,然后真空浓缩,得到的固体。固体用乙酸乙酯和石油醚作为冲洗液进行柱色谱分离2次,真空蒸干溶剂。将固体烘干后,获得中间体化合物m1 8.1g,收率85%,lcms(esi离子源)测得m/z:277.0。
[0069]
其他的荧蒽类中间体按照以上方式进行合成,只是采用的原料有所差异,合成实施例2
–
8的原料如表1所示。
[0070]
表1
[0071][0072]
合成实施例9
[0073]
化合物a2的合成
[0074][0075]
在三口烧瓶中,加入a2-1(10g,22.5mmol),a2-2(6.2g,222.5mmol),三(二亚苄基丙酮)二钯(pd2(dba)3,(0.4g,0.45mmol),三叔丁基膦t-bu3p(0.5g,2.25mmol),叔丁醇钠naobu-t,(6.5g,67.6mmol),在氮气氛围下在100ml甲苯溶剂(toluene)中搅拌,反应液升温至110℃,搅拌反应3小时。反应液冷却至室温,加入50ml水萃取分相,甲苯相蒸干,固体用甲苯-石油醚进行柱色谱分离纯化,然后真空浓缩,浓缩液用甲苯进行重结晶纯化,得到化合物a2 11g,收率72%,lcms(esi离子源)测得m/z:684.3,1hnmr(dmso-d6):δ,8.02
–
7.87(m,1h),7.80
–
7.68(m,4h),7.63
–
7.52(m,4h),7.51
–
7.43(m,3h),7.43
–
7.20(m,7h),7.19
–
7.12(m,2h),7.05
–
6.90(m,1h),2.84(t,2h),2.70(m,3h),2.17(m,2h),2.07
–
1.87(m,2h),1.73
–
1.25(m,14h)。
[0076]
采用反应通式3的实施例操作方法与化合物a2完全相同,只是原料进行替换,合成实施例10
–
21合成化合物的原料和收率等如表2所示。
[0077]
表2
[0078][0079]
上述合成实施例制备的化合物1h nmr如表3所示。
[0080]
表3
[0081]
[0082][0083]
合成实施例22
[0084]
化合物b43的合成:
[0085][0086]
在三口烧瓶,依次加入b43-1(10g,38mmol)、b43-2(9g,38mmol)、k2co3(13g,114mmol)、四(三苯基膦)钯pd(pph3)4(0.87g,0.76mmol)再加入80ml甲苯中,10ml水,10ml乙醇的混合溶剂中,反应液升温至回流,搅拌保持6小时。反应液冷却后,加入100ml盐水萃取,有机相用无水硫酸钠干燥,然后真空浓缩,得到的固体用甲苯和石油醚作为冲洗液进行柱色谱分离。然后再进行甲苯重结晶2次得到固体,固体烘干,获得化合物b43 12g,收率72%;lcms(esi离子源)测得m/z:421.1;1h nmr(dmso-d6):δ8.68
–
8.32(m,1h),8.07(d,2h),7.71
–
7.63(m,3h),7.63
–
7.58(m,2h),7.58
–
7.53(m,1h),7.52
–
7.37(m,4h),7.23(m,2h),3.46(s,4h).
[0087]
采用反应通式2的其他实施例与化合物b43完全相同,只是原料进行替换,合成实施例23
–
35合成化合物的原料和收率等如表4所示。
[0088]
表4
[0089][0090]
上述合成实施例制备的化合物1h nmr如表5所示。
[0091]
表5
25g,收率51%,lcms(esi离子源)测得m/z:649.2。
[0097]
化合物d39的合成:在三口烧瓶中,加入d39-3(10g,15.4mmol),d39-4(1.7g,15.4mmol),三(二亚苄基丙酮)二钯(pd2(dba)3,(0.13g,0.15mmol),三叔丁基膦t-bu3p(0.33g,1.54mmol),叔丁醇钠naobu-t,(4.4g,46.2mmol),在氮气氛围下在甲苯溶剂(toluene)中搅拌,反应液升温至110℃,搅拌反应3小时。反应液冷却至室温,用甲苯和水萃取,甲苯相蒸干,固体用甲苯-石油醚进行柱色谱分离纯化,然后真空浓缩,浓缩液用甲苯进行重结晶纯化,得到化合物d397.8g,收率70%;lcms(esi离子源)测得m/z:725.2;1hnmr(dmso-d6):δ9.02(s,1h),8.14
–
8.08(m,1h),8.04
–
7.97(m,2h),7.74(m,1h),7.65(s,1h),7.63
–
7.30(m,18h),7.30
–
7.25(m,2h),7.21(m,2h),3.45(s,4h).
[0098]
采用反应通式4的其他实施例与合成实施例36相同,只是原料有所差别,合成实施例37-40的原料和收率等如表6所示。
[0099]
表6
[0100][0101]
上述合成实施例制备的化合物1h nmr如表7所示。
[0102]
表7
[0103][0104][0105]
合成实施例41
[0106]
化合物d12的合成
[0107][0108]
化合物d12-3的合成:在三口烧瓶中,加入d12-1(20g,47.7mmol),d12-2(15.5g,47.7mmol),三(二亚苄基丙酮)二钯(pd2(dba)3,(0.4g,0.95mmol),三叔丁基膦t-bu3p(1.04g,4.8mmol),叔丁醇钠naobu-t(13.7g,143.2mmol),在氮气氛围下在200ml甲苯溶剂(toluene)中搅拌,反应液升温至110℃,搅拌反应3小时。反应液冷却至室温,用甲苯和水萃取,甲苯相蒸干,固体用甲苯-石油醚进行柱色谱分离纯化,然后真空浓缩,浓缩液用甲苯正己烷混合溶剂进行重结晶纯化,得到d12-3化合物23.1g,收率73%。lcms(esi离子源)测得m/z:662.1。
[0109]
化合物d12-4的合成:在三口烧瓶,依次加入d12-3(20g,30.1mmol、k2co3(12.4g,90.5mmol)、醋酸钯pd(oac)2(0.06g,0.30mmol)、三环己基膦四氟硼酸盐pcy3-hbf4(0.11g,3mmol),再加入200mln,n-二甲基乙酰胺(dma)中,反应液升温至130℃,搅拌反应8小时。反应结束后冷却后,真空浓缩,剩余的溶液用甲苯和石油醚作为冲洗液进行柱色谱分离纯化。收集的液体真空蒸干后,用甲苯进行重结晶,得到固体,烘干,获得中间体化合物d12-4 13.5g,收率72%。lcms(esi离子源)测得m/z:627.2。
[0110]
化合物d12的合成:在三口烧瓶中,加入d12-4(10g,15.9mmol),d12-5(2.5g,15.9mmol),三(二亚苄基丙酮)二钯(pd2(dba)3,(0.27g,0.32mmol),三叔丁基膦t-bu3p(0.34g,1.59mmol),叔丁醇钠naobu-t,(4.6g,47.9mmol),在氮气氛围下在甲苯溶剂(toluene)中搅拌,反应液升温至110℃,搅拌反应3小时。反应液冷却至室温,用甲苯和水萃取,甲苯相蒸干,固体用甲苯-石油醚进行柱色谱分离纯化,然后真空浓缩,浓缩液用甲苯进行重结晶纯化,得到化合物d12 8.4g,收率75%;lcms(esi离子源)测得m/z:703.2;1h nmr(dmso-d6):δ9.21
–
9.04(m,2h),8.53(m,1h),8.09
–
8.01(m,2h),7.96
–
7.86(m,2h),7.84
–
7.79(m,2h),7.76
–
7.66(m,3h),7.64
–
7.58(m,1h),7.57
–
7.38(m,13h),7.06(d,2h),6.07(s,2h).
[0111]
采用反应通式5的其他实施例与合成实施例41相同,只是原料有所差别,合成实施例42-44的原料和收率等如表8所示。
[0112]
表8
[0113][0114]
上述实施例化合物的1h nmr如9所示。
[0115]
表9
[0116][0117]
如图1所示的底发光器件指光从tft基板侧出光,如图2所示的顶发光器件指光从远离tft基板侧出光,顶发光器件与底发光器件相比,在阴极上有一层覆盖层,器件发光颜色可以是红色、绿色、蓝色、白色、黄色等各种颜色。
[0118]
一些实施方式中,底发光器件的构造可选以下任意结构:
[0119]
①
阳极/空穴注入层/空穴传输层1/发光层/电子传输层1/阴极;
[0120]
②
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/阴极;
[0121]
③
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/电子传输层2/阴极;
[0122]
④
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/电子传输层2/电子注入层/阴极;
[0123]
⑤
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/电子传输层2/多层阴极;
[0124]
⑥
阳极/空穴注入层/空穴传输层1/发光层1/载流子产生层/空穴传输层2/发光层2/电子传输层1/阴极;
[0125]
⑦
阳极/空穴注入层/空穴传输层1/发光层1/载流子产生层/空穴传输层2/发光层2/电子传输层1/电子传输层2/阴极;
[0126]
⑧
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层1/载流子产生层/空穴传输层3/发光层2/电子传输层1/阴极;
[0127]
⑨
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层1/载流子产生层/空穴传输层3/发光层2/电子传输层1/电子传输层2/阴极。
[0128]
一些实施方式中,顶发光器件的构造可选以下任意结构:
[0129]
①
阳极/空穴注入层/空穴传输层1/发光层/电子传输层1/阴极/覆盖层;
[0130]
②
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/阴极/覆盖层;
[0131]
③
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/电子传输层2/阴极/覆盖层;
[0132]
④
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层/电子传输层1/电子传输层2/电子注入层/阴极/覆盖层;
[0133]
⑤
阳极/空穴注入层/空穴传输层1/空穴传输层1/发光层/电子传输层1/电子传输层2/多层阴极/覆盖层;
[0134]
⑥
阳极/空穴注入层/空穴传输层1/发光层1/载流子产生层/空穴传输层2/发光层
2/电子传输层1/阴极/覆盖层;
[0135]
⑦
阳极/空穴注入层/空穴传输层1/发光层1/载流子产生层/空穴传输层2/发光层2/电子传输层1/电子传输层2/阴极/覆盖层;
[0136]
⑧
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层1/电子传输层2/载流子产生层/空穴传输层3/发光层2/电子传输层1/阴极/覆盖层;
[0137]
⑨
阳极/空穴注入层/空穴传输层1/空穴传输层2/发光层1/电子传输层3/载流子产生层/空穴传输层3/发光层2/电子传输层1/电子传输层2/阴极/覆盖层。
[0138]
上述任意器件结构中,空穴传输层1厚度一般为40
–
150nm,常采用含芳基胺化合物,可以采用芳单胺,也可以采用芳香多胺,要求具有高的空穴迁移率,能够降低驱动电压,玻璃转化温度超过100℃,避免高温下结晶。空穴传输层a可选自以下分子结构:空穴传输层2和空穴传输层3厚度一般为3
–
150nm。
[0139]
发光材料层中主体材料(host)的含量大于发光客体掺杂材料(dopant),可选发光材料层中客体掺杂材料的质量百分比为1%
–
20%。客体掺杂材料可以包括磷光材料、或荧光材料、或热激活延迟荧光材料,红光、绿光、蓝光可从以上三种客体掺杂材料选择。
[0140]
发光主体可以选择一种发光主体或者两种发光主体。
[0141]
电子传输层用于将阴极的电子传输至发光层,一般采用具有空穴传输能力的材料,厚度为10
–
50nm。
[0142]
覆盖层形成于器件的半透明阴极远离基板一侧,能够使光输出效率提高,厚度一般为50
–
90nm,例如50nm、55nm、57nm、59nm、62nm、64nm、67nm、68nm、70nm、75nm、77nm、79nm、80nm、82nm、85nm、88nm、90nm等,在450
–
650nm之间的光透过率≥65%,例如68%、69%、73%、77%、79%、83%、88%、93%等。
[0143]
本发明的上述荧蒽类化合物可以用在oled底发光和顶发光器件的空穴传输层、发光层、电子传输层以及顶发光的覆盖层,针对不同的功能层给出器件实施例,具体如下所述。
[0144]
器件实施例1
[0145]
本实施例采用荧蒽类化合物a2作为空穴传输层1 104的材料,以蓝光器件(底发光)制作方法为例,制备工艺为:
[0146]
在玻璃材质的基底101上,形成透明阳极ito膜层,膜厚150nm,得到第一电极102作为阳极,之后蒸镀与空穴传输材料的混合材料作为空穴注入层103,混合比例为3:97(质量比),之后蒸镀100nm厚度本发明化合物得到第一层空穴传输层104,然后蒸镀20nm厚度化合物得到
第二层空穴传输层105,然后以95:5的蒸镀速率蒸镀发光主体材料与发光体材料30nm,制作蓝光发光单元106,然后蒸镀10nm形成空穴阻挡层107,然后以4:6的蒸镀速率蒸镀与形成厚度30nm的电子传输层108,之后形成厚度100nm的镁银(质量比1:9),作为第二电极109。
[0147]
器件实施例2
–
8分别用上述荧蒽类化合物a6、a17、a26、a42、a50、a59、a67替代器件实施例1的a2,对比例1采用化合物(e1)替代器件实施例1的a2,其他材料保持不变。器件测试采用keithley电源,ms-75光谱辐射计的组合测试设备,电压和效率采用10ma/cm2时的电压(v)和电流效率表示(单位为cd/a),寿命为10ma/cm2电流时亮度衰减为初始亮度的95%所需要的时间,具体如表10所示。
[0148]
表10
[0149][0150]
器件实施例9
[0151]
本实施例采用荧蒽类化合物c1作为电子传输层108材料,以蓝光器件(底发光)制作方法为例,制备工艺为:
[0152]
在玻璃材质的基底101上,形成透明阳极ito膜层,膜厚150nm,得到第一电极102作
为阳极,之后蒸镀与空穴传输材料的混合材料作为空穴注入层103,混合比例为3:97(质量比),之后蒸镀100nm厚度的化合物得到第一层空穴传输层104,然后蒸镀20nm厚度化合物得到第二层空穴传输层105,然后以95:5的蒸镀速率蒸镀发光主体材料与发光体材料30nm,制作蓝光发光单元106,然后蒸镀10nm形成空穴阻挡层107,然后以4:6的蒸镀速率蒸镀本发明的化合物c1与形成厚度30nm的电子传输层108,之后形成厚度100nm的镁银(质量比1:9),作为第二电极109。
[0153]
器件实施例10
–
14分别用本发明的化合物c11、c27、c36、c38、c39替代器件实施例9的c1,对比例2采用化合物(e2)替代器件实施例1的c1,其他材料保持不变。器件测试采用keithley电源,ms-75光谱辐射计的组合测试设备,电压和效率采用10ma/cm2时的电压(v)和电流效率表示(单位为cd/a),寿命为10ma/cm2电流时亮度衰减为初始亮度的95%所需要的时间,具体如表11所示。
[0154]
表11
[0155][0156][0157]
器件实施例15
[0158]
本实施例采用荧蒽类化合物b1作为顶发光器件的覆盖层110材料,以蓝光器件制作方法为例,制备工艺为:
[0159]
在玻璃材质的基底101上,形成不透光的反射阳极ag/ito膜层,膜厚为100nm/15nm,得到第一电极102作为阳极,之后蒸镀与空穴传输材料的混合材料作为空穴注入层103,混合比例为3:97(质量比),之后蒸镀100nm厚度的化合物得到第一层空穴传输层104,然后蒸镀20nm厚度化合物得到第二层空穴传输层105,然后以95:5的蒸镀速率蒸镀发光主体材料与发光体材料30nm,制作蓝光发光单元106,然后蒸镀10nm形成空穴阻挡层107,然后以4:6的蒸镀速率蒸镀化合物
与形成厚度30nm的电子传输层108,之后形成厚度15nm的镁银(质量比1:9),作为第二电极109,之后蒸镀厚度为70nm本发明的化合物形成的覆盖层110。
[0160]
器件实施例16
–
21分别用本发明化合物b3、b9、b17、b31、b43、b47替代器件实施例15的b1,对比例3采用化合物(e3)替代器件实施例1的b1,其他材料保持不变。器件测试采用keithley电源,ms-75光谱辐射计的组合测试设备,电压和效率采用10ma/cm2时的电压(v)和电流效率与色坐标ciey的比值表示(单位为cd/a/ciey),折射率是采用椭圆偏振仪在设定测试波长为460nm时,测试材料单层膜,膜厚为70nm获得,具体如表12所示。
[0161]
表12
[0162][0163]
一些实施方式中,发光主体材料采用多种材料混合制成,其中发光掺杂材料采用的金属铱化合物选自但不限于以下化合物:
[0164][0165][0166]
器件实施例22
[0167]
本实施例采用荧蒽类化合物c7作为红光磷光发光层主体材料,以底发光器件制作方法为例,其中红光发光层由发光主体材料和磷光发光材料构成,以金属铱化合物ir1作为发光掺杂材料,制备工艺为:在玻璃材质的基底101上,形成透明阳极ito膜层,膜厚150nm,得到第一电极102作为阳极,之后蒸镀与空穴传输材料的混合材料作为空穴注入层103,混合比例为3:97(质量比),之后蒸镀100nm厚度的化合物得到第一层空穴传输层104,然后蒸镀100nm厚度
的化合物得到第二层空穴传输层105,然后以48:49:3的蒸镀速率蒸镀40nm本发明化合物c7d1与ir1的混合材料,制作红光发光单元106,然后蒸镀10nm形成空穴阻挡层107,然后蒸镀与混合比例为4:6(质量比)形成厚度30nm的电子传输层108,之后形成厚度100nm的镁银(质量比1:9),作为第二电极109。
[0168]
器件实施例23
–
34分别用本发明化合物c7:d5(含量比38:59)、c13:d12(含量比48:49)、c13:d25(含量比59:38)、c7:d32(含量比48:49)、c21:d39(含量比48:49)、c26:d48(含量比38:59)、c26:d57(含量比48:49)、c26:d69(含量比48:49)、c7:对比化合物5(含量比48:49)、对比化合物4:d39(含量比48:49)、d73、d81以及对比化合物4:对比化合物5(含量比48:49)、对比化合物6替代器件实施例22的c7:d1(含量比48:49)作为发光层105,其中对比化合物4、5和6的化学结构分别为:器件测试采用keithley电源,ms-75光谱辐射计的组合测试设备,电压和效率采用10ma/cm2时的电压(v)和电流效率表示(单位为cd/a),寿命为10ma/cm2电流时亮度衰减为初始亮度的95%所需要的时间,如表13所示。
[0169]
表13
[0170][0171][0172]
热稳定性实验
[0173]
在一个模拟生产蒸镀的腔体中,此腔体具有真空系统、加热系统以及速率探测系统,腔体抽真空至10-7torr,按照的蒸镀速率加热材料,经过持续加热200小时后,将腔体打开,取出装材料容器中的材料,通过hplc测试中剩余的材料的纯度,其中对比化合物7、8和9的化学结构分别为通过比较未加热前的初始的材料纯度以及加热200小时后的材料纯度,确认材料耐高温的情况,如表14所示。
[0174]
表14
[0175]
材料名称材料的初始纯度(hplc)200小时后的纯度(hplc)a299.992%99.981%对比化合物799.994%99.963%c199.993%99.962%对比化合物899.992%99.944%d2599.992%99.992%对比化合物999.994%99.981%
[0176]
从表14可以看出,本发明的化合物相比不带烷基的化合物具有更好的耐热稳定性。
[0177]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其发明构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。